Prostate cancer is the second-leading cause of cancer-related deaths among men in the United States [
1]. This warrants a large body of research to decipher the intricate biology of the disease and devise effective treatments. The hallmark of prostate cancer progression is the androgen receptor (AR) signaling pathway, which is mainly responsible for prostate cell survival, proliferation, and resistance to treatment. Despite significant advancements in the targeting of this pathway, the emergence of castration-resistant prostate cancer (CRPC) and its inherent resistance to conventional anti-androgen therapies have underscored the complexity of the disease. Metastatic CRPC (mCRPC), under pressure from regimens targeting the AR pathway, can undergo molecular changes driving resistance to these treatment regimens [
2]. A pivotal facet of this resistance arises from the alternative splicing of AR mRNA, generating AR splice variants (AR-Vs) [
3]. AR-Vs are truncated isoforms of the AR that do not bind the androgens but have the potential to be constitutively active. Among the AR-Vs, AR-V7 is the most researched splice variant. AR-V7 lacks the ligand binding domain (LBD), which allows it to activate the AR signaling pathway in the absence of androgens [
4]. Suppression of ligand-mediated AR-FL signaling results in increased AR-V7 expression in CRPC cell line models [
5]. AR-V7 activates a distinct expression signature enriched for cell cycle genes without requiring the presence of androgens or functional AR-FL [
5], though AR-FL continues to be expressed in higher abundance [
4,
5,
6]. This shift toward AR-V7-mediated signaling after suppression of ligand-dependent AR-FL is considered an important mechanism contributing to drug resistance to CRPC therapy [
5].
2. Prostate Cancer Treatment Landscape
Traditional therapeutic strategies for treating prostate cancer involve a sequential progression through available treatment modalities in response to evolving disease status. Upon the initial diagnosis of localized prostate cancer, often detected through early screening, interventions encompass local treatments, including surgery and radiation. About one-third of patients with localized disease experience recurrence following local therapies [
11]. Disease recurrence is often indicated by prostate-specific antigen (PSA) (i.e., biochemical recurrence) or imaging. Those encountering a biochemical or local recurrence might continue under observation, bypassing ADT to mitigate its potential side effects, until reaching a predetermined threshold, subsequently leading to the adoption of ADT in the form of luteinizing hormone-releasing hormone (LHRH) antagonists or agonists. This regimen may persist, with intermittent periods of treatment holidays, until signs of disease progression via PSA, imaging, or clinical symptoms become apparent [
12]. While these initial treatments can yield remission for patients—terminology that is handled cautiously in prostate cancer oncology—most individuals will eventually develop diseases resistant to ADT, that is, CRPC.
In cases where the malignancy has extended beyond its original site before local treatments, the healthcare provider and the patient need to make a judicious decision about the initiation of androgen-deprivation therapy (ADT) either as a singular agent, in combination with radiation for low-volume metastatic disease [
13], or with docetaxel for both low- and high-volume metastatic disease [
14].
In instances where a patient receives an initial diagnosis of metastatic prostate cancer (de novo mHSPC), the standard approach involves the prompt initiation of ADT with anti-androgen therapies such as bicalutamide, abiraterone, or enzalutamide plus or minus docetaxel [
15]. Over time, patients may exhibit signs of progression as their tumor tissue develops resistance to AR-targeted therapies. Consequently, alternative therapeutic avenues, including poly (ADP-ribose) polymerase (PARP) inhibitors, chemotherapy, radium 223, radiation therapy, prostate-specific membrane antigen (PSMA)-targeted treatments, and immunotherapy, are explored [
12].
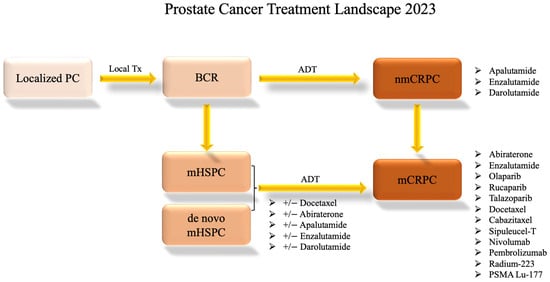
Figure 2 shows this progression through treatments.
Figure 2. Prostate Cancer Systemic Treatment Landscape. This flow chart shows the standard progression through available treatment options for varying disease statuses as of 2023. Clinical trial options are not included. The information presented is subject to change pending future drug approvals. ADT: androgen-deprivation therapy; BCR: biochemical recurrence; local Tx: local treatment such as radiation or surgery; mCRPC: metastatic castration-resistant prostate cancer; mHSPC: metastatic hormone-sensitive prostate cancer; nmCRPC: non-metastatic castration-resistant prostate cancer; PC: prostate cancer.
3. Drugs That Target AR-Vs Directly
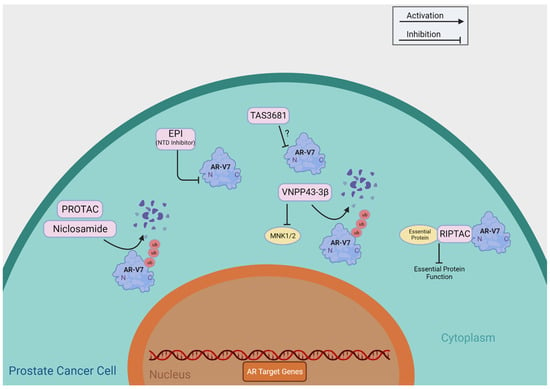
Table 1 summarizes agents having putative roles in suppressing AR-Vs through direct functional inhibition, including niclosamide, TAS3681, EPI-7386, Au-AR pep-PROTAC, VNPP433-3β, and RIPTAC. Researchers provide a detailed review of these agents currently undergoing clinical development and summarize their putative mechanisms of action in Figure 3.
Figure 3. Direct Targeting of AR-V7. This figure provides a brief overview of the mechanisms of action of the compounds that directly target AR-V7, as shown in Table 1. ?: The mechanism of action of TAS3681 is yet to be published.
3.1. Niclosamide
Within the landscape of existing therapies and compounds undergoing clinical trials, several avenues hold promise for directly targeting AR-Vs. A notable contender is niclosamide, which exhibits potent inhibitory effects on AR-V7. Niclosamide’s mechanism of action for prostate cancer is expected to involve hindering AR-V7 recruitment to androgen-responsive elements such as the PSA promoter and curbing its protein expression by instigating degradation [
16]. Furthermore, preclinical data suggest this compound demonstrates proficiency in curbing prostate cancer cell proliferation in vitro and mitigating tumor growth in vivo [
16]. Notably, niclosamide not only overcomes resistance to enzalutamide but also synergizes with enzalutamide therapy, thus portraying its potential utility in advanced prostate cancer cases, particularly those resistant to enzalutamide [
16].
The therapeutic trajectory of niclosamide encountered hurdles in 2018 during a Phase I dose-escalation study testing oral niclosamide plus standard-dose enzalutamide in men with mCRPC previously treated with abiraterone (NCT02532114). The trial revealed that the doses needed to attain efficacious plasma concentrations yielded substantial toxicity, precluding the viability of niclosamide as an oral anticancer agent. Consequently, the trial was prematurely halted due to futility, casting uncertainty over the future development of niclosamide as a cancer therapy due to its poor pharmacokinetic properties [
17].
Fortunately, further research has been conducted to establish the maximum tolerable dose and recommended dosage of niclosamide. A Phase Ib trial investigating reformulated niclosamide in tandem with abiraterone acetate and prednisone in men with CRPC elicited findings that underscored the combination’s favorable tolerability profile and the achievement of therapeutic niclosamide concentrations (NCT02807805). Encouragingly, select patients recorded noteworthy declines in PSA levels, coupled with radiographic ameliorations. The findings suggested that the combination of niclosamide and abiraterone is clinically active [
18]. Phase II of this trial is ongoing.
A recent investigation has endeavored to amend niclosamide’s poor pharmaceutical properties by analyzing the efficacy of a novel series of niclosamide analogs and further characterizing the structure-activity relationship of the compound. Several analogs exhibited equivalent or improved anti-proliferation effects in cell lines, potent AR-V7 downregulation, and improved metabolic activity [
19]. Further investigation into the clinical use of these compounds may yield exciting results for the field.
3.2. TAS3681
TAS3681 is a promising experimental agent that may have dual capabilities to act against androgens and downregulate AR-FL and AR-Vs. In in vitro and in vivo experiments, TAS3681 has shown effectiveness in prostate cancer cell lines, including those with both normal and alternatively spliced AR, such as AR-V7 [
20]. Before entering clinical trials, preclinical research revealed that TAS3681 hinders the movement of AR into the cell nucleus after binding and lowers the levels of AR-FL and AR-V7 [
21]. Its potential to combat AR-V7-positive cancer lines through AR downregulation prompted the initiation of a Phase I study for patients with mCRPC (NCT02566772). The ongoing trial investigates the safety, tolerability, and efficacy of the compound through a dose-escalation phase followed by an expansion phase that enrolled patients who progressed on either abiraterone or enzalutamide +/− taxane-based chemotherapy. This study is ongoing, but some preliminary results from the dose-escalation phase published in 2021 demonstrated that TAS3681 has a well-tolerated safety profile and exhibits anticancer effects in patients with mCRPC at 300 mg twice daily [
22]. The results from this trial are sure to yield intriguing insights into the practicality of TAS3681 as a compound with the potential to inhibit both AR-FL and AR-V7, though it was designed to target AR-FL.
3.3. EPI Compounds
Considering the indispensable role of the androgen-receptor N-terminal domain (AR-NTD), which contains the most transcriptional activity of AR across androgen levels, this domain becomes an attractive target for novel therapeutic interventions. EPI compounds have been shown to inhibit AR transcriptional activity, degrade AR-FL and AR-Vs, and prevent AR-V-associated resistance [
23]. In 2010, investigators identified EPI-001, a small molecule that blocked the transactivation of AR-NTD. It interacted with the AF-1 region of AR-NTD, disrupting protein-protein interactions and reducing the binding of ARs to androgen-response elements on target genes. Importantly, EPI-001 effectively blocked androgen-induced cancer cell proliferation and caused a reduction in CRPC tumor growth without causing any noticeable toxicity in mouse models [
24]. EPI-001 is a mixture of four stereoisomers, with the most potent stereoisomer being EPI-002. EPI-001 and EPI analogs covalently bound to the NTD, effectively blocking AR and its splice variant’s (ARv567es) transcriptional activity, and showed reduced CRPC tumor growth in experiments [
25].
EPI-506, a prodrug of EPI-002, emerged as a potential therapeutic approach targeting both full-length AR and AR splice variants responsible for resistance to prevailing treatments [
26]. However, a Phase I/II investigation involving men with mCRPC was terminated due to a lack of efficacy and poor bioavailability (NCT02606123). Investigation into another EPI compound, EPI-7386, demonstrated improved pharmaceutical properties [
27]. It is now in Phase I/II testing as a monotherapy (NCT04421222) and in combination with enzalutamide (NCT05075577) [
28].
3.4. PROTAC
A new drug called proteolysis-targeting chimera (PROTAC) has been designed to degrade AR-FL and AR-V7 in a specific manner using LBD, or the DNA binding domain (DBD), and an MDM2-dependent mechanism. The PROTAC agents that target AR-LBD, such as ARV-110 [
29], are considered ineffective against AR-Vs since the variants lack that domain. The PROTAC molecules that target the DBD, however, may be effective against AR-Vs. To improve the effectiveness of PROTAC, the drug is delivered using an ultra-small gold-peptide complex platform. The resulting drug, Au-AR pep-PROTAC, efficiently degrades AR-FL and AR-V7 in prostate cancer cells, leading to tumor regression in both sensitive and resistant mouse models [
30]. Clinical investigations into this compound have yet to be initiated.
3.5. VNPP433-3β
VNPP433-3β is a galeterone derivative recently developed to concurrently target AR and MNK1/2. Despite its successful progression in Phase 2 clinical trials for the treatment of mCRPC, galeterone encountered challenges of early trial termination due to high censorship for the primary endpoint during the pivotal Phase 3 clinical trial (NCT02438007) [
31]. As a result, researchers have developed a galeterone-derivative drug candidate, VNPP433-3β, effective against CRPC by targeting AR-FL and AR-Vs. VNPP433-3β promotes the degradation of full-length AR and AR-V7 while also inhibiting MNK1/2, leading to reduced phosphorylation of eIF4E and mRNA translation. Preclinical investigation with cell lines (CWR22Rv1 and LNCaP) on this novel drug compound has shown promising results [
32].
3.6. RIPTAC
Regulated Induced Proximity Targeting Chimera (RIPTAC) is another novel compound currently being investigated to target ARs. RIPTACs are compounds designed to selectively bind a protein that is overproduced in cancer cells. In the case of prostate cancer, the ideal candidate is AR, which is overexpressed as castration resistance occurs. After the compound binds AR, its mechanism comes into play. It is then able to selectively bind and therefore sequester an effector protein (EP) essential to cell function and viability. The cell dies off as a result if enough of the EP is incorporated in the AR:RIPTAC:EP stable ternary complex [
33]. There must be significantly elevated levels of AR in the cell in order for enough essential proteins to be sequestered for the cell to die off. Because of this, RIPTAC-mediated disruption of EP should only affect cancerous cells that are producing AR at far above normal levels. Preclinical investigations have demonstrated the ability of AR-RIPTAC to successfully target and kill prostate cancer cells [
33]. The company Halda has assayed the ability of RIPTACs to form ternary complexes with an undisclosed EP and AR in VCaP cells, and selective apoptosis in AR-high cells was observed. This indicates that RIPTACs will indeed be able to directly target AR-V7, which is expressed in VCaP. Additionally, in VCaP xenograft mouse models, tumor growth inhibition and tumor regression were demonstrated [
34]. Future studies may reveal interesting information about this drug as it transitions to IND filing, which is slated for 2024 [
33].
Table 1. Compounds that Directly Target AR-Vs. The compounds are listed along with the prostate cancer models used in preclinical studies, AR-V7 target genes tested to assess inhibitors’ activities, their mechanism of action, NCT numbers, and associated references. AR-V7 target genes tested include both the actual gene expressions and the promoters of indicated genes conjugated to a luciferase reporter. N/A: the expression of AR-V7 target genes or reporter assays using gene promoters were not tested in the cited references. *: The cited references were conference abstracts in which experimental details were not provided. This table was adapted and expanded from a section of Table 3 in the Kanayama et al. review [
3].