Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Oxidative stress, characterized by an imbalance between the production of reactive oxygen species (ROS) and the cellular anti-oxidant defense mechanisms, plays a critical role in the pathogenesis of various human diseases. ROS are either produced through cellular processes or environmental factors. Of note, oxidative stress has been described as a secondary effect within the pathology of several rare monogenic diseases and sometimes been called a common denominator.
- oxidative stress
- reactive oxygen species (ROS)
- cellular redox balance
1. The Mitochondrial Respiratory Chain
The mitochondrial respiratory chain, or electron transport chain (ETC), is one of the biggest endogenous sources of ROS in eukaryotic cells which use aerobic metabolism (Figure 1) [1][2]. In humans, the enzyme complexes I (NADH:ubiquinone oxidoreductase), II (succinate:ubiquinone oxidoreductase), and III (ubiquinol:cytochrome c oxidoreductase) are somewhat “leaky”, leading to direct one-electron transfer to molecular oxygen and the perpetual production of the superoxide anion radical (O2·−) as side reactions [2][3].
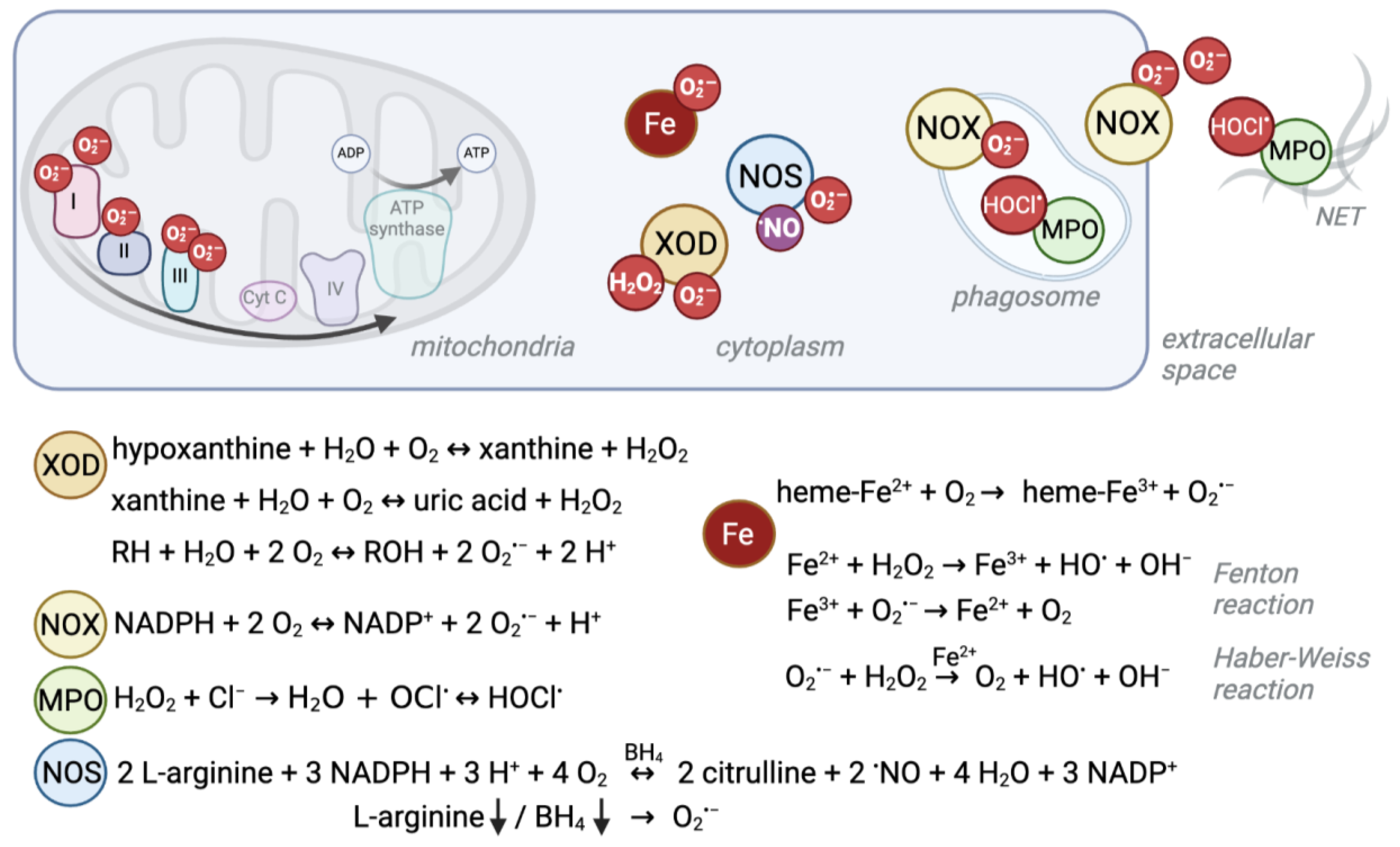
Figure 1. Schematic overview of ROS production sites and reactions. Several enzymatic reactions as well as interactions of iron with oxygen give rise to reactive oxygen species. Their localization and production level can differ between cell compartments and cell types. For example, red blood cells experience high levels of superoxide due to their heme-bound iron (Fe2+) and oxygen interaction, whereas the mitochondrial electron transport chain is a major source of ROS in other cells. Under ischemic conditions, xanthine oxidase (XOD) produces superoxide anions (O2·−) and hydrogen peroxide (H2O2) in the cytosol. NADPH oxidases (NOX) mainly produce O2·− to kill pathogens in the phagosome or extracellular space. Similarly, myeloperoxidase (MPO) produces hypochlorous acid (HOCl.) from H2O2 which derives from NOX-produced O2·− in phagosomes and neutrophil extracellular traps (NETs). Instead, nitric oxide synthases (NOS) normally produce nitric oxide (.NO) as a signaling molecule. Depletion of its substrate arginine or cofactor (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4) can cause the enzyme to uncouple and produce O2·−.
Complex I is encoded by more than 40 genes [4], complex II by four genes [5] and complex III by 11 genes [6]. However, the specific sites and levels of ROS production within the different protein complexes depend on the respiration substrate, whether the cell experiences norm- or hypoxic conditions, and on the inhibitor used in experimental setups [7]. Furthermore, respiratory complexes can assemble into supercomplexes, which decreases ROS production [8]. Astrocytes contain high amounts of free complex I and thus potentially higher levels of ROS compared to neurons, which display complex I and III assembly [9]. Thus, the diversity of conditions as well as of cell type specificities make it often hard to pin down the specific set of genes of respiratory complex subunits which involve pathomechanisms related to ROS production in humans. This is also exemplified by the fact that some pathogenic mutations within subunits do not lead to ROS elevation, as shown for H2O2 in a mouse model for Leigh syndrome [10]. By contrast, in other experiments, mutations of genes involved in complex I, II, or III formation were indeed shown to create greater “leakiness”, and cause elevated ROS levels and oxidative stress [4][11][12][13].
Since the ETC generates a strong proton gradient used for oxidative phosphorylation, transport processes, and heat production, it can be difficult to disentangle whether the pathophysiological mechanism triggered by mutation of ETC genes is based on compromised primary functions, or elevated ROS levels. However, at least for complex I deficiency, elevated ROS production and its consequent oxidative damage were shown to induce apoptotic molecular pathways leading to neuron de.generation and hence neurological symptoms, the main features of the disorder [11].
Deficiencies in respiratory complexes have similar but wide-ranging symptoms from neonatal death, lactic acidosis, myopathy, hepatopathy, encephalopathy, Leigh syndrome [14], Leber hereditary optic neuropathy (LHON) [15], to adult-onset neurological symptoms such as some forms of Parkinson disease [16][17]. Isolated complex I deficiency is the most prevalent genetic disorder of oxidative phosphorylation [18].
As outlined above, it is challenging to disentangle the underlying factors, but the numerous potential ROS production sites and levels might be part of the explanation of the broad range of symptoms and disorder severities.
2. Heme in Red Blood Cells
Another ROS formation hotspot, despite being devoid of mitochondria, is the cytosol of red blood cells (RBCs). RBCs transport oxygen from the lung to peripheral tissues and superoxide can be formed when O2 interacts with the iron (Fe2+) of the heme group of hemoglobin (Hb) [19]. Spontaneous autoxidation results in O2·− and methemoglobin (Hb-Fe3+, MetHb, Hb M) (Figure 1) [18][20], and ca. 1% of all Hb is present as Hb M in healthy individuals [21].
This normally occurring rate of superoxide production can be exceeded by pathogenic genetic variants that cause conformational changes to the globin chain proteins that contain heme as a prosthetic group. This phenomenon is well described for, e.g., autosomal recessive sickle cell disease (SCD) (Table 1). SCD is the most common severe hemoglobinopathy worldwide and is caused by a missense pathogenic variant in the globin beta-chain (HBB), resulting in Hb S (NM_000518.5(HBB):c.20A>T (p.Glu7Val); E6V) [22], which is unstable, prone to polymerization and autoxidation [23]. Besides other ROS sources like iron release, this autoxidation is the primary source for oxidative stress in sickle cells, and it leads to loss of membrane structure and function and consequent multisystem disease [24][25][26][27]. When iron is released, it impacts the oxidative balance through transfer of single electrons via the Fenton reaction (Fe2+ + H2O2 → Fe3+ + HO· + OH−) and the Haber–Weiss reaction (O2·− + H2O2 → O2 + HO· + OH− catalyzed by iron) (Figure 1) [28].
Table 1. Summary of discussed genes and phenotypes. AR, autosomal recessive, AD, autosomal dominant, XLR, X-linked recessive, OMIM®, Online Mendelian Inheritance in Man database.
| Protein | Gene (OMIM® no.) | Monogenic Disease (OMIM®no.) | Inheritance | Described by |
|---|---|---|---|---|
| ROS production | ||||
| Hemoglobin beta-locus | HBB (141900) | Sickle cell disease (603903) | AR | [22] |
| Methemoglobinemia, beta type (617971) | AD | [21] | ||
| Cytochrome b(-245), beta subunit, p91-phox | CYBB (300481) | Chronic granulomatous disease (306400) | XLR | [29] |
| Immunodeficiency 34, mycobacteriosis (300645) | XLR | [30] | ||
| Cytochrome b(-245), alpha subunit, p22-phox | CYBA (608508) | Chronic granulomatous disease 4 (233690) | AR | [31] |
| Neutrophil cytosolic factor-1, p47-phox | NCF1 (608512) | Chronic granulomatous disease 1 (233700) | AR | [32] |
| Neutrophil cytosolic factor-2, p67-phox | NCF2 (608515) | Chronic granulomatous disease 2 (233710) | AR | [33] |
| Neutrophil cytosolic factor-4, p40-phox | NCF4 (601488) | Chronic granulomatous disease 3 (613960) | AR | [34] |
| Myeloperoxidase | MPO (606989) | Myeloperoxidase deficiency (254600) | AR | [35] |
| Xanthine dehydrogenase | XDH (607633) | Xanthinuria, type I (278300) | AR | [36] |
| Nitric oxide synthase 1 | NOS1 (163731) | - | ||
| Nitric oxide synthase 2 | NOS2 (163730) | - | ||
| Nitric oxide synthase 3 | NOS3 (163729) | - | ||
| enzymatic ROS clearance | ||||
| superoxide dismutase 1 | SOD1 (147450) | Amyotrophic lateral sclerosis 1 (105400) | AD, AR | [37] |
| Spastic tetraplegia and axial hypotonia, progressive (618598) | AR | [38] | ||
| superoxide dismutase 2 | SOD2 (147460) | - | ||
| superoxide dismutase 3 | SOD3 (185490) | - | ||
| catalase | CAT (115500) | Acatalasemia (614097) | AR | [39] |
| glutathione peroxidase 1 | GPX1 (138320) | Hemolytic anemia due to glutathione peroxidase deficiency (614164) | AR | [40] |
| glutathione peroxidase 4 | GPX4 (138322) | Spondylometaphyseal dysplasia, Sedaghatian type (250220) | AR | [41] |
| glutathione peroxidase 2, 3, 5-8 | GPX2 (138319) GPX3 (138321) GPX5 (603435) GPX6 (607913) GPX7 (615784) GPX8 (617172) | - | ||
| peroxiredoxin 3 | PRDX3 (604769) | Corneal dystrophy, punctiform, and polychromatic pre-Descemet (619871) | AD | [42] |
| Spinocerebellar ataxia, autosomal recessive 32 (619862) | AR | [43] | ||
| peroxiredoxin 1, 2, 4, 5, 6 | PRDX1 (176763) PRDX2 (600538) PRDX4 (300927) PRDX5 (606583) PRDX6 (602316) | - | ||
| thiols and thiol production | ||||
| gamma-glutamylcysteine synthetase, catalytic subunit | GCLC (606857) | Hemolytic anemia due to gamma-glutamylcysteine synthetase deficiency (230450) | AR | [44] |
| glutathione synthetase | GSS (601002) | Glutathione synthetase deficiency (266130) | AR | [45] |
| Hemolytic anemia due to glutathione synthetase deficiency (231900) | AR | [46] | ||
| thioredoxin 2 | TXN2 (609063) | Combined oxidative phosphorylation deficiency 29 (616811) | AR | [47] |
| thioredoxin | TXN (187700) | - | ||
| thiol recycling | ||||
| glutathione reductase | GSR (138300) | Hemolytic anemia due to glutathione reductase deficiency (618660) | AR | [48] |
| thioredoxin reductase 1, 2, 3 | TXNRD1 (601112) TXNRD2 (606448) TXNRD3 (606235) | - | ||
| NADPH production | ||||
| glucose 6P-dehydrogenase | G6PD (305900) | Hemolytic anemia, G6PD deficient (favism) (611162) | XL | [49] |
| 6-phosphogluconate dehydrogenase | PGD (172200) | Phosphogluconate dehydrogenase deficiency (619199) | AD | [50] |
| malic enzyme 1 | ME1 (154250) | - | ||
| malic enzyme 3 | ME3 (604626) | - | ||
| glutamate dehydrogenase 1 | GLUD1 (138130) | Hyperinsulinism-hyperammonemia syndrome (606762) | AD | [51] |
| glutamate dehydrogenase 2 | GLUD2 (300144) | - | ||
| isocitrate dehydrogenase 2 | IDH2 (147650) | D-2-hydroxyglutaric aciduria 2 (613657) | AD | [52] |
| isocitrate dehydrogenase 1 | IDH1 (147700) | - | ||
| methylenetetrahydrofolate dehydrogenase 1 | MTHFD1 (172460) | Combined immunodeficiency and megaloblastic anemia with or without hyperhomocysteinemia (617780) | AR | [53] |
| methylenetetrahydrofolate dehydrogenase 2 | MTHFD2 (604887) | - | ||
| 10-formyltetrahydrofolate dehydrogenase (mitochondrial) | ALDH1L2 (613584) | neuro-ichthyotic syndrome? | AR | [54] |
| 10-formyltetrahydrofolate dehydrogenase (cytosolic) | ALDH1L1 (600249) | - | ||
Also, other HBB variants have been described to cause methemoglobinemia. For example, the so-called Hb M-Hyde Park (Hb M-Akita) (NM_000518.4(HBB):c.277C>T (p.His93Tyr)) variant leads to conformational changes at the heme binding site and hence higher rates of methemoglobin formation. Inheritance is autosomal dominant [21][55].
3. NADPH Oxidases and Myeloperoxidase
Besides these cellular processes in which ROS are formed passively as byproducts, the cell can actively produce high levels of ROS through dedicated enzymes. NADPH oxidases (NOX) use reduced nicotinamide adenine dinucleotide phosphate (NADPH) to generate superoxide (Figure 1) [56]. NOX play an important role in the innate host defense. For the so-called “respiratory burst”, superoxide is released into the extracellular space or phagosomes to fight off pathogenic bacteria or fungi [57]. Furthermore, NOX enzymes were found to produce intracellular ROS at lower levels, which is believed to serve signaling functions [58] and to control the cellular redox balance by oxidizing NADPH and through ROS formation [59]. The importance of the ability to generate high ROS levels is exemplified by the detrimental consequences of NOX disruption.
Several monogenic disorders are related to NOX subunits. The genes that encode the NOX2 complex in phagocytes are related to chronic granulomatous disease [60][61]: CYBB pathogenic variants cause X-linked recessive chronic granulomatous disease (CGDX, [29]) and immunodeficiency 34 [30]), CYBA pathogenic variants cause autosomal recessive (AR) CGD4 [31], NCF1 pathogenic variants cause AR CGD1 [32], NCF2 pathogenic variants cause AR CGD2 [33], and NCF4 pathogenic variants cause AR CGD3 [34] (Table 1). These rare primary immunodeficiencies increase susceptibility to life-threatening bacterial and fungal infections and lead to development of granulomas [62].
Pathogenic variants in other NADPH oxidases were shown to increase susceptibility to inflammatory bowel disease (NOX1 and DUOX2, [63]), or cause congenital hypothyroidism (DUOX2, Thyroid dyshormonogenesis 6, AR, [64]; DUOXA2, Thyroid dyshormonogenesis 5, AR, [65], Table 1). The latter one is the result of disrupted H2O2 production through mutated DUOX2, which would be required for organification of iodide for thyroid hormone synthesis catalyzed by thyroid peroxidase [61][66].
Another enzyme of the innate immune response is the heme-containing enzyme myeloperoxidase (MPO, Table 1, Figure 1). It is highly abundant in azurophilic granules of neutrophils [67] and is a crucial component in neutrophil extracellular nets [68]; it is also found—to a lesser extent—in monocytes [69]. To kill pathogens, MPO creates strongly reactive species, such as hypochlorous acid (HOCl.) from hydrogen peroxide (H2O2) and chloride (Cl−) [70][71]. Autosomal recessive MPO deficiency is the most common phagocyte defect but is asymptomatic in the majority of patients, suggesting compensatory mechanisms [72]. However, higher susceptibility to fungal infections, especially Candida albicans, has been described [35]. This vulnerability might become apparent only in combination with comorbidities like diabetes, which itself increases the risk of infections [71]. Similarly, a recent study described a case of partial DiGeorge syndrome together with MPO deficiency. DiGeorge syndrome is characterized by immunodeficiency among other symptoms. However, the patient had more frequent and severe infections than expected for partial DiGeorge alone, which the authors explained by co-occurrence of MPO deficiency [73]. On the other hand, a protective mechanism against cardiovascular disease by absence of the potentially oxidative stress causing MPO enzyme has also been discussed [72].
4. Other ROS Sources
In addition to the above-described reactions and enzymes, there are additional sources that can produce significant amounts of reactive species under certain physiological circumstances.
For example, xanthine oxidoreductase (XDH/XOD), encoded by the XDH gene (Table 1, Figure 1), exists in two interconvertible isoforms. Both forms utilize hypoxanthine or xanthine to produce uric acid. However, the predominant form has xanthine dehydrogenase (XDH) activity, and uses NAD+ as cofactor to produce NADH, whereas the xanthine oxidase (XOD) form uses oxygen to produce the superoxide anion and H2O2 [74]. XDH can be converted to XOD by irreversible limited proteolysis or reversibly by thiol oxidation (reviewed in [75]), e.g., in hypoxic/ischemic tissue [76]. Homozygous or compound heterozygous pathogenic XDH variants cause type I xanthinuria (XAN1) [36]. The symptoms, low serum and urine uric acid and xanthinuria leading to urolithiasis [77], result from XDH’s primary function.
Furthermore, nitric oxide synthases (NOS1-3) can become sources of O2·− (Figure 1). Normally, they use L-arginine to produce nitric oxide (·NO), which belongs to the group of reactive nitrogen species (RNS) and serves as an important signaling molecule, especially for the vascular tone [78]. However, persistent oxidative stress, and thus reduced levels of the cofactor (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4), lead to uncoupling of endothelial NOS (eNOS, NOS3) so that the enzyme produces O2·− instead of ·NO [79]. Furthermore, O2·− is produced by NOS2 in arginine-depleted macrophages [80]. Although some NOS3 variants have been described to increase the risk for certain conditions like pregnancy- induced hypertension [81] or ischemic stroke [82], no clear monogenic disorder has been described for NOS1, NOS2, or NOS3.
At lower amounts, ROS can also be byproducts of other enzymes like cytochrome P450 or cyclooxygenase (reviewed in [83]).
Environmental factors that create elevated ROS levels are, e.g., UV radiation, ionizing radiation, smoking and air pollution, chemicals such as drugs, and certain types of food like the fava bean [84][85]. Often, genetic defects in the anti-oxidative machinery remain unnoticed until such environmental stresses hit and overwhelm the residual anti-oxidative capacity of the cell, e.g., in hemolytic anemia due to glucose 6-phosphate dehydrogenase (G6PDH) deficiency (see below).
This entry is adapted from the peer-reviewed paper 10.3390/biom14020206
References
- Muller, F. The Nature and Mechanism of Superoxide Production by the Electron Transport Chain: Its Relevance to Aging. J. Am. Aging Assoc. 2000, 23, 227–253.
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344.
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial Complex II and Reactive Oxygen Species in Disease and Therapy. Redox Rep. 2020, 25, 26–32.
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial Respiratory Complex I Dysfunction Promotes Tumorigenesis through ROS Alteration and AKT Activation. Hum. Mol. Genet. 2011, 20, 4605–4616.
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The Genetic Basis of Isolated Mitochondrial Complex II Deficiency. Mol. Genet. Metab. 2020, 131, 53–65.
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome bc1 Complex. Science 1998, 281, 64–71.
- Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415.
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480.
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068.
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions That Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. 2019, 30, 824–832.e3.
- Perier, C.; Tieu, K.; Guégan, C.; Caspersen, C.; Jackson-Lewis, V.; Carelli, V.; Martinuzzi, A.; Hirano, M.; Przedborski, S.; Vila, M. Complex I Deficiency Primes Bax-Dependent Neuronal Apoptosis through Mitochondrial Oxidative Damage. Proc. Natl. Acad. Sci. USA 2005, 102, 19126–19131.
- Tretter, L.; Sipos, I.; Adam-Vizi, V. Initiation of Neuronal Damage by Complex I Deficiency and Oxidative Stress in Parkinson’s Disease. Neurochem. Res. 2004, 29, 569–577.
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of Reactive Oxygen Species by the Mitochondrial Electron Transport Chain. J. Neurochem. 2002, 80, 780–787.
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh Syndrome: One Disorder, More than 75 Monogenic Causes. Ann. Neurol. 2016, 79, 190–203.
- Riordan-Eva, P.; Harding, A.E. Leber’s Hereditary Optic Neuropathy: The Clinical Relevance of Different Mitochondrial DNA Mutations. J. Med. Genet. 1995, 32, 81–87.
- Di Monte, D.A. Mitochondrial DNA and Parkinson’s Disease. Neurology 1991, 41, 38–42; discussion 42–43.
- Ikebe, S.; Tanaka, M.; Ozawa, T. Point Mutations of Mitochondrial Genome in Parkinson’s Disease. Brain Res. Mol. Brain Res. 1995, 28, 281–295.
- Tucker, E.J.; Compton, A.G.; Calvo, S.E.; Thorburn, D.R. The Molecular Basis of Human Complex I Deficiency. IUBMB Life 2011, 63, 669–677.
- Misra, H.P.; Fridovich, I. The Generation of Superoxide Radical during the Autoxidation of Hemoglobin. J. Biol. Chem. 1972, 247, 6960–6962.
- Winterbourn, C.C. Oxidative Reactions of Hemoglobin. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1990; Volume 186, pp. 265–272.
- Schnedl, W.J.; Queissner, R.; Schenk, M.; Enko, D.; Mangge, H. Hereditary Methemoglobinemia Caused by Hb M-Hyde Park (Hb M-Akita) (HBB:c.277C > T; p.His93Tyr). Wien. Klin. Wochenschr. 2019, 131, 381–384.
- Mears, J.G.; Lachman, H.M.; Cabannes, R.; Amegnizin, K.P.; Labie, D.; Nagel, R.L. Sickle Gene. Its Origin and Diffusion from West Africa. J. Clin. Investig. 1981, 68, 606–610.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-Cell Disease. Lancet 2010, 376, 2018–2031.
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated Autoxidation and Heme Loss due to Instability of Sickle Hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241.
- Hebbel, R.P.; Ney, P.A.; Foker, W. Autoxidation, Dehydration, and Adhesivity May Be Related Abnormalities of Sickle Erythrocytes. Am. J. Physiol. 1989, 256, C579–C583.
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608.
- Orrico, F.; Laurance, S.; Lopez, A.C.; Lefevre, S.D.; Thomson, L.; Möller, M.N.; Ostuni, M.A. Oxidative Stress in Healthy and Pathological Red Blood Cells. Biomolecules 2023, 13, 1262.
- Papanikolaou, G.; Pantopoulos, K. Iron Metabolism and Toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211.
- Zurro, N.B.; Tavares de Albuquerque, J.A.; França, T.T.; Vendramini, P.; Arslanian, C.; Tavares-Scancetti, F.; Condino-Neto, A. A Novel Mutation in CYBB Gene in a Patient with Chronic Colitis and Recurrent Pneumonia due to X-Linked Chronic Granulomatous Disease. Pediatr. Blood Cancer 2018, 65, e27382.
- Bustamante, J.; Picard, C.; Fieschi, C.; Filipe-Santos, O.; Feinberg, J.; Perronne, C.; Chapgier, A.; de Beaucoudrey, L.; Vogt, G.; Sanlaville, D.; et al. A Novel X-Linked Recessive Form of Mendelian Susceptibility to Mycobaterial Disease. J. Med. Genet. 2007, 44, e65.
- Teimourian, S.; Zomorodian, E.; Badalzadeh, M.; Pouya, A.; Kannengiesser, C.; Mansouri, D.; Cheraghi, T.; Parvaneh, N. Characterization of Six Novel Mutations in CYBA: The Gene Causing Autosomal Recessive Chronic Granulomatous Disease. Br. J. Haematol. 2008, 141, 848–851.
- Curnutte, J.T.; Berkow, R.L.; Roberts, R.L.; Shurin, S.B.; Scott, P.J. Chronic Granulomatous Disease due to a Defect in the Cytosolic Factor Required for Nicotinamide Adenine Dinucleotide Phosphate Oxidase Activation. J. Clin. Investig. 1988, 81, 606–610.
- Patiño, P.J.; Rae, J.; Noack, D.; Erickson, R.; Ding, J.; de Olarte, D.G.; Curnutte, J.T. Molecular Characterization of Autosomal Recessive Chronic Granulomatous Disease Caused by a Defect of the Nicotinamide Adenine Dinucleotide Phosphate (reduced Form) Oxidase Component p67-Phox. Blood 1999, 94, 2505–2514.
- van de Geer, A.; Nieto-Patlán, A.; Kuhns, D.B.; Tool, A.T.; Arias, A.A.; Bouaziz, M.; de Boer, M.; Franco, J.L.; Gazendam, R.P.; van Hamme, J.L.; et al. Inherited p40phox Deficiency Differs from Classic Chronic Granulomatous Disease. J. Clin. Investig. 2018, 128, 3957–3975.
- Lehrer, R.I.; Cline, M.J. Leukocyte Myeloperoxidase Deficiency and Disseminated Candidiasis: The Role of Myeloperoxidase in Resistance to Candida Infection. J. Clin. Investig. 1969, 48, 1478–1488.
- Tsukasa, K.; Toshihiro, N.; Motoshi, K.; Tatsuo, H.; Kusuki, N. Biochemical Studies on the Purine Metabolism of Four Cases with Hereditary Xanthinuria. Clin. Chim. Acta 1984, 137, 189–198.
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 Mutations Associated with Amyotrophic Lateral Sclerosis Analysis of Variant Severity. Sci. Rep. 2022, 12, 103.
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 Deficiency: A Novel Syndrome Distinct from Amyotrophic Lateral Sclerosis. Brain 2019, 142, 2230–2237.
- Takahara, S. Progressive Oral Gangrene Probably due to Lack of Catalase in the Blood (acatalasaemia); Report of Nine Cases. Lancet 1952, 2, 1101–1104.
- Necheles, T.F.; Maldonado, N.; Barquet-Chediak, A.; Allen, D.M. Homozygous Erythrocyte Glutathione-Peroxidase Deficiency: Clinical and Biochemical Studies. Blood 1969, 33, 164–169.
- Sedaghatian, M.R. Congenital Lethal Metaphyseal Chondrodysplasia: A Newly Recognized Complex Autosomal Recessive Disorder. Am. J. Med. Genet. 1980, 6, 269–274.
- Henríquez-Recine, M.A.; Marquina-Lima, K.S.; Vallespín-García, E.; García-Miñaur, S.; Benitez Del Castillo, J.M.; Boto de Los Bueis, A. Heredity and in Vivo Confocal Microscopy of Punctiform and Polychromatic Pre-Descemet Dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 1661–1667.
- Rebelo, A.P.; Eidhof, I.; Cintra, V.P.; Guillot-Noel, L.; Pereira, C.V.; Timmann, D.; Traschütz, A.; Schöls, L.; Coarelli, G.; Durr, A.; et al. Biallelic Loss-of-Function Variations in PRDX3 Cause Cerebellar Ataxia. Brain 2021, 144, 1467–1481.
- Konrad, P.N.; Richards, F., 2nd; Valentine, W.N.; Paglia, D.E. -Glutamyl-Cysteine Synthetase Deficiency. A Cause of Hereditary Hemolytic Anemia. N. Engl. J. Med. 1972, 286, 557–561.
- Jellum, E.; Kluge, T.; Börresen, H.C.; Stokke, O.; Eldjarn, L. Pyroglutamic Aciduria—A New Inborn Error of Metabolism. Scand. J. Clin. Lab. Investig. 1970, 26, 327–335.
- Mohler, D.N.; Majerus, P.W.; Minnich, V.; Hess, C.E.; Garrick, M.D. Glutathione Synthetase Deficiency as a Cause of Hereditary Hemolytic Disease. N. Engl. J. Med. 1970, 283, 1253–1257.
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; Kobayashi, S.; Terrile, C.; Wolf, P.; Schaper, J.; et al. Human Thioredoxin 2 Deficiency Impairs Mitochondrial Redox Homeostasis and Causes Early-Onset Neurodegeneration. Brain 2016, 139, 346–354.
- Loehr, G.W.; Waller, H.D. A new enzymopenic hemolytic anemia with glutathione reductase deficiency. Med. Klin. 1962, 57, 1521–1525.
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069.
- Brewer, G.J.; Dern, R.J. A new inherited enzymatic deficiency of human erythrocytes: 6-phosphogluconate dehydrogenase deficiency. Am. J. Hum. Genet. 1964, 16, 472–476.
- Brandt, A.; Agarwal, N.; Giri, D.; Yung, Z.; Didi, M.; Senniappan, S. Hyperinsulinism Hyperammonaemia (HI/HA) Syndrome due to GLUD1 Mutation: Phenotypic Variations Ranging from Late Presentation to Spontaneous Resolution. J. Pediatr. Endocrinol. Metab. 2020, 33, 675–679.
- Kranendijk, M.; Struys, E.A.; van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; van der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; de Klerk, J.B.; et al. IDH2 Mutations in Patients with D-2-Hydroxyglutaric Aciduria. Science 2010, 330, 336.
- Watkins, D.; Schwartzentruber, J.A.; Ganesh, J.; Orange, J.S.; Kaplan, B.S.; Nunez, L.D.; Majewski, J.; Rosenblatt, D.S. Novel Inborn Error of Folate Metabolism: Identification by Exome Capture and Sequencing of Mutations in the MTHFD1 Gene in a Single Proband. J. Med. Genet. 2011, 48, 590–592.
- Sarret, C.; Ashkavand, Z.; Paules, E.; Dorboz, I.; Pediaditakis, P.; Sumner, S.; Eymard-Pierre, E.; Francannet, C.; Krupenko, N.I.; Boespflug-Tanguy, O.; et al. Deleterious Mutations in ALDH1L2 Suggest a Novel Cause for Neuro-Ichthyotic Syndrome. NPJ Genom. Med. 2019, 4, 17.
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858.
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH Oxidases: An Overview from Structure to Innate Immunity-Associated Pathologies. Cell. Mol. Immunol. 2014, 12, 5–23.
- Iles, K.E.; Forman, H.J. Macrophage Signaling and Respiratory Burst. Immunol. Res. 2002, 26, 95–105.
- Nauseef, W.M. Biological Roles for the NOX Family NADPH Oxidases. J. Biol. Chem. 2008, 283, 16961–16965.
- Schröder, K. NADPH Oxidases: Current Aspects and Tools. Redox Biol 2020, 34, 101512.
- Matute, J.D.; Arias, A.A.; Wright, N.A.M.; Wrobel, I.; Waterhouse, C.C.M.; Li, X.J.; Marchal, C.C.; Stull, N.D.; Lewis, D.B.; Steele, M.; et al. A New Genetic Subgroup of Chronic Granulomatous Disease with Autosomal Recessive Mutations in p40phox and Selective Defects in Neutrophil NADPH Oxidase Activity. Blood 2009, 114, 3309–3315.
- O’Neill, S.; Brault, J.; Stasia, M.-J.; Knaus, U.G. Genetic Disorders Coupled to ROS Deficiency. Redox. Biol. 2015, 6, 135–156.
- de Oliveira-Junior, E.B.; Bustamante, J.; Newburger, P.E.; Condino-Neto, A. The Human NADPH Oxidase: Primary and Secondary Defects Impairing the Respiratory Burst Function and the Microbicidal Ability of Phagocytes. Scand. J. Immunol. 2011, 73, 420–427.
- Hayes, P.; Dhillon, S.; O’Neill, K.; Thoeni, C.; Hui, K.Y.; Elkadri, A.; Guo, C.H.; Kovacic, L.; Aviello, G.; Alvarez, L.A.; et al. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 489–502.
- Moreno, J.C.; Bikker, H.; Kempers, M.J.E.; van Trotsenburg, A.S.P.; Baas, F.; de Vijlder, J.J.M.; Vulsma, T.; Ris-Stalpers, C. Inactivating Mutations in the Gene for Thyroid Oxidase 2 (THOX2) and Congenital Hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102.
- Zamproni, I.; Grasberger, H.; Cortinovis, F.; Vigone, M.C.; Chiumello, G.; Mora, S.; Onigata, K.; Fugazzola, L.; Refetoff, S.; Persani, L.; et al. Biallelic Inactivation of the Dual Oxidase Maturation Factor 2 (DUOXA2) Gene as a Novel Cause of Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 605–610.
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of Two Human Thyroid cDNAs Encoding New Members of the NADPH Oxidase Family. J. Biol. Chem. 2000, 275, 23227–23233.
- Schultz, J.; Kaminker, K. Myeloperoxidase of the Leucocyte of Normal Human Blood. I. Content and Localization. Arch. Biochem. Biophys. 1962, 96, 465–467.
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase Is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood 2011, 117, 953–959.
- Bos, A.; Wever, R.; Roos, D. Characterization and Quantification of the Peroxidase in Human Monocytes. Biochim. Biophys. Acta 1978, 525, 37–44.
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-Mediated Regulation of Innate and Adaptive Immunity: The Role of Myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817.
- van der Veen, B.S.; de Winther, M.P.J.; Heeringa, P. Myeloperoxidase: Molecular Mechanisms of Action and Their Relevance to Human Health and Disease. Antioxid. Redox Signal. 2009, 11, 2899–2937.
- Kutter, D.; Devaquet, P.; Vanderstocken, G.; Paulus, J.M.; Marchal, V.; Gothot, A. Consequences of Total and Subtotal Myeloperoxidase Deficiency: Risk or Benefit ? Acta Haematol. 2000, 104, 10–15.
- Abraitytė, S.; Kotsi, E.; Devlin, L.A.; Edgar, J.D.M. Unexpected Combination: DiGeorge Syndrome and Myeloperoxidase Deficiency. BMJ Case Rep. 2020, 13, e232741.
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine Dehydrogenase/xanthine Oxidase and Oxidative Stress. Age 1997, 20, 127–140.
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian Xanthine Oxidoreductase—Mechanism of Transition from Xanthine Dehydrogenase to Xanthine Oxidase. FEBS J. 2008, 275, 3278–3289.
- McCord, J.M. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N. Engl. J. Med. 1985, 312, 159–163.
- Sebesta, I.; Stiburkova, B.; Krijt, J. Hereditary Xanthinuria Is Not so Rare Disorder of Purine Metabolism. Nucleosides Nucleotides Nucleic Acids 2018, 37, 324–328.
- Förstermann, U. Nitric Oxide and Oxidative Stress in Vascular Disease. Pflug. Arch. 2010, 459, 923–939.
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace. Circulation 2006, 113, 1708–1714.
- Xia, Y.; Zweier, J.L. Superoxide and Peroxynitrite Generation from Inducible Nitric Oxide Synthase in Macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958.
- Arngrímsson, R.; Hayward, C.; Nadaud, S.; Baldursdóttir, A.; Walker, J.J.; Liston, W.A.; Bjarnadóttir, R.I.; Brock, D.J.; Geirsson, R.T.; Connor, J.M.; et al. Evidence for a Familial Pregnancy-Induced Hypertension Locus in the eNOS-Gene Region. Am. J. Hum. Genet. 1997, 61, 354–362.
- Berger, K.; Stögbauer, F.; Stoll, M.; Wellmann, J.; Huge, A.; Cheng, S.; Kessler, C.; John, U.; Assmann, G.; Ringelstein, E.B.; et al. The glu298asp Polymorphism in the Nitric Oxide Synthase 3 Gene Is Associated with the Risk of Ischemic Stroke in Two Large Independent Case-Control Studies. Hum. Genet. 2007, 121, 169–178.
- Gwozdzinski, K.; Pieniazek, A.; Gwozdzinski, L. Reactive Oxygen Species and Their Involvement in Red Blood Cell Damage in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2021, 2021, 6639199.
- Tavassolifar, M.J.; Vodjgani, M.; Salehi, Z.; Izad, M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020, 2020, 5793817.
- Kattamis, C.A.; Kyriazakou, M.; Chaidas, S. Favism: Clinical and Biochemical Data. J. Med. Genet. 1969, 6, 34–41.
This entry is offline, you can click here to edit this entry!