Heart failure (HF) is marked by dampened cardiac contractility. A mild therapeutic target that improves contractile function without desensitizing the β-Adregenric system during HF may improve cardiac contractility and potentially survival. Inhibiting PKCα activity may fit the criteria of the therapeutic target with milder systemic effects that still boosts contractility in HF patients. PKCα activity has been observed to increase during HF. This increase in PKCα activity is perplexing because it is also accompanied by the up-regulation of a molecular braking mechanism.
- myocardial infarction
- translocation
- lipid
- contractile dysfunction
- β-adrenergic system desensitization
1. Introduction
Heart failure (HF) is a progressive condition and is marked by reduced output which in turn, evokes an increase in catecholamines and other neuroendocrine factors [1]. These factors can activate the reactive signaling pathways thus negatively influencing the heart and further down-regulating its functional performance [2]. Through enhancing the cardiac output, the persistent negative feedback loop could be disrupted as it may lead to a secondary reduction in neuroendocrine drive. Inotropes seem to be chemical agents of choice for this as they can selectively enhance the cardiac contractility [3][4][5][6][7][8]. Unfortunately, these agents are associated with high mortality rates, possibly due to the desensitization of the entire β-adrenergic system [4][5][6][7][8]. Thus, in many clinical settings, the safety of traditional inotropes is controversial [4][5][6][7][8]. Therefore, there is a critical need for heart failure research to focus on finding therapeutics that are milder alternatives to positive inotropes and do not come with a high risk of β-adrenergic system desensitization [8][9][10][11][12]. The overall clinical need for selectively enhancing cardiac contractility as a therapeutic strategy remains unresolved. Enhancing just the precise amount of contractility within physiological limits may increase the survival propensity of patients during HF.
Clinically, diastolic heart failure (DHF) and systolic heart failure (SHF) are the most common forms of heart failure (HF) disease. These syndromes are marked by a progressive loss in contractility, ejection failure, ventricular chamber dilation, ventricular wall thinning, increased peripheral vascular resistance and dysregulated fluid homeostasis [1][2][3][4]. The propensity towards these forms of cardiac insufficiency could be linked to the activation of PKCα molecule in DHF & SHF conditions [11][12]. The PKCα molecule act as a nodal integrator of cardiac contractility and may act as a refined and milder alternative of positive inotropes to effectively boost contractility in HF [11][12][13][14][15][16][17][18]. Clinical evidence suggests the positive inotropes worsens the prognosis in individuals with somewhat stable HF as they are susceptible to enhance contractility beyond safety limits [4-8]. Inhibiting PKCα may provide a more refined and milder target possibility which might augment cardiac contractility within the physiological limits. Observations show that loss of PKCα activity could lead to a 20-30% increase in contractility during HF [10][11][12][13][14][15][16][17][18]. Experimental observations in angiotensin II (Ang II)-stimulated cardiomyocytes show that, in part, HF associated contractile dysfunction can be regulated through the Gαq/DAG/PKCα/DGKζ signaling cascade [19][20][21]. PKCα belongs to the conventional protein kinase C (cPKC) family of serine/threonine protein kinases. These kinases are canonically activated by Ca+2 and lipid signaling [11]. PKCα is the most prominent member of the PKC family and is expressed in mouse, human and rabbit heart tissue [10][11][12]. Previous observations have linked PKCα to impaired left ventricular filling and ejection during heart failure. PKCα is necessary and sufficient to induce ventricular systolic and diastolic dysfunction [10][11]. Pharmacological and genetic inhibition of PKCα clearly improves contractility during heart failure, attenuating the extent of damage and disease [10][11]. Prolonged activation of PKCα may lead to serious malignant outcomes by inducing systolic and diastolic dysfunction [9][10][11][12][13][14][15][16][17].
DGKζ is another key functional effector that signals during Gαq-induced heart failure [15]. Previous research has shown that cardiac-specific overexpression of DGKζ suppresses remodeling and fibrosis in the left ventricle (LV) independent of hemodynamic regulation. Thus, overexpression of DGKζ under these conditions rescues angiotensin-induced congestive heart failure [15]. Previous data indicate that overexpression of DGKζ also improves survival after MI [16]. Human heart failure is linked to increased PKCα activity [14]. This linkage is surprising, as PKCα activity in most cell types is exquisitely regulated [22][23]. PKCα activity in cardiomyocytes is also tightly regulated through a molecular braking mechanism. As soon as PKCα is activated, the molecular brakes in place to check this increase in PKCα concentration should also kick in, acting on the common activator. This molecular braking mechanism begs the question: why is PKCα activity increased and maintained during heart failure despite the concurrent activation of a braking mechanism? Here, I propose an explanation based on data analyzing a local DAG signaling cascade that may be responsible for the increase in PKCα activity during HF.
Human heart failure is linked to increased PKCα activity [12]. This linkage is surprising, as PKCα activity in most cell types is exquisitely regulated [22][23]. PKCα activity in cardiomyocytes is also tightly regulated through a molecular braking mechanism. As soon as PKCα is activated, the molecular brakes in place to check this increase in PKCα concentration should also kick in, acting on the common activator. This molecular braking mechanism begs the question: why is PKCα activity increased and maintained during heart failure despite the concurrent activation of a braking mechanism? Here, I propose an explanation based on data analyzing a local DAG signaling cascade that may be responsible for the increase in PKCα activity during HF.
Here, I propose DAG signaling homeostasis during Ang II-induced heart failure can be regulated through a molecular loop between the positive and negative effector molecules PKCα and DGKζ. This proposed molecular loop may improve understanding of the molecular mechanisms involved in the complex spatiotemporal organization of DAG-PKCα- DGKζ signaling during post-MI cardiac remodeling events [29-34][22][23][24][25][26][27][28]. Using a computational model, this study shows the proposed molecular loop has a dual regulatory character. During basal conditions, a net negative feedback loop may prevail and regulate local DAG concentration. Interestingly, upon stimulation conditions, a positive feedback effect on DAG signaling is observed. This positive feedback can possibly explain the link between persistently high DAG levels and malignant outcomes. The transition from negative to positive feedback depends on the local biosynthesis rate of DAG and, in turn, on the mutual interactions between positive and negative DAG effector molecules.
2. Results and Discussions
2.1 Local DAG Signaling Regulation in Cardiomyocytes
The model I propose in Figure 1 & 2, describe local regulation of DAG homeostasis in cardiomyocytes The model is composed of two molecular components: 1. PKCα, the target protein of DAG signaling, can exist in one of four states: cytosolic dormant (PKCIIα), inactive membrane (PKCIα), active membrane (PKCIαA) or active cytosolic (PKCIIαA). 2. DGKζ, the attenuator protein of DAG signaling, can be in one of three states: cytosolic (DGKIIζ), active membrane (DGKIζ), or phosphorylated/inactive membrane (DGKIζP). Both these components migrate to the plasma membrane in a DAG-dependent manner. Once in the plasma membrane, DGKζ forms complex C1 with PKCα. These two components interact in a closed loop and regulate DAG homeostasis in a negative feedback loop.
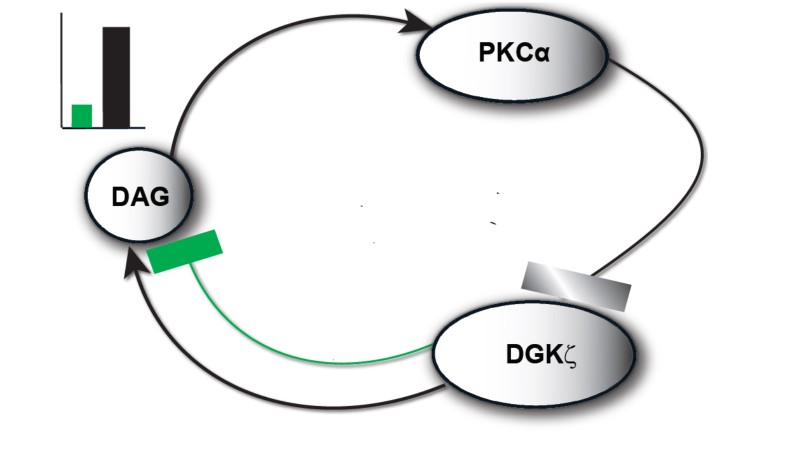
Figure 1. The regulatory molecular loop between the target (PKCα) and attenuator (DGKζ) of local DAG signaling.
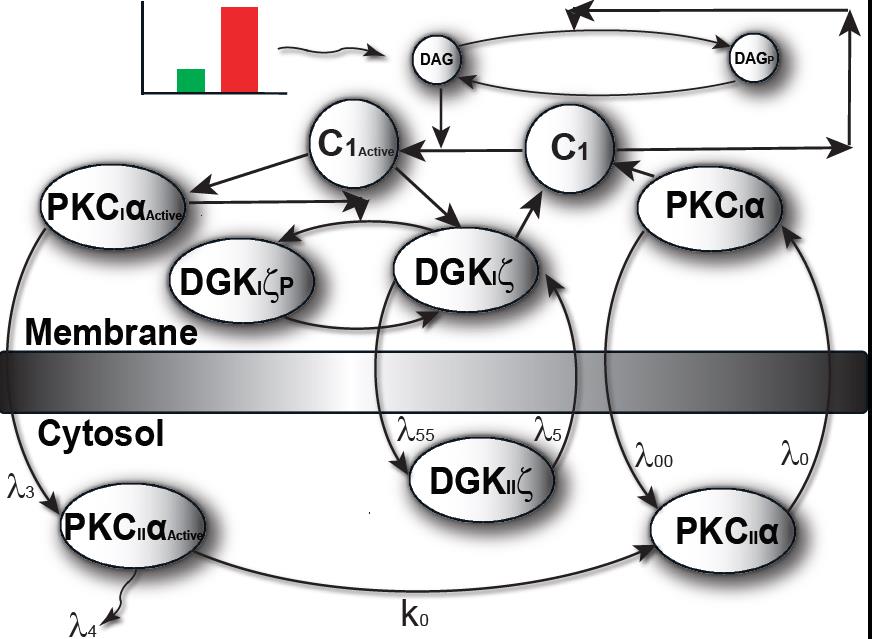
Figure 2. The two-compartment regulatory model of local DAG signaling in cardiomyocytes. Here, one of the compartments is cytosol, whereas the other compartment is the plasma membrane.
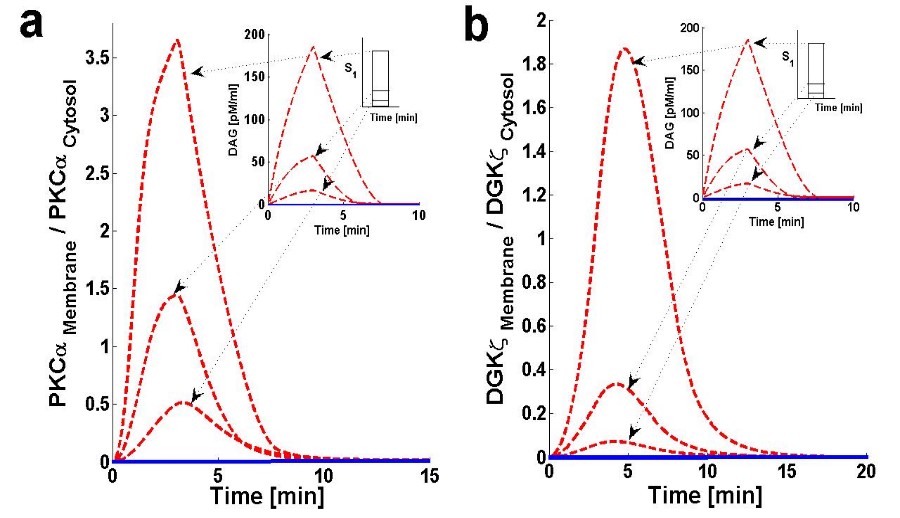
Figure 3. Membrane to cytosol (M/C) ratio of target & attenuator molecules of DAG signaling.
Using the computation model, I next determined the translocation characteristics of both the target and attenuator molecules of DAG signaling in cardiomyocytes (Figure 3). In the model, translocation characteristics were determined by measuring the membrane to the cytosol (M/C) ratio of target and attenuator molecules. The M/C ratio describes the relative distribution of PKCα and DGKζ molecules in the membrane and cytosolic compartments. For PKCα, this ratio is an indirect index of activation. According to the proposed model, the M/C ratio for DGKζ could be indirectly linked to kinase deactivation. A higher M/C ratio indicates high rates of molecular migration from the cytosol to the plasma membrane Ang II-induced biosynthesis of DAG was implemented using a three-minute pulse, as described in the previous section. The strength of the pulse is described by an arbitrary parameter S1. Here, the parameter S1 is set at three arbitrary levels: 0.5,2 and 6. In the absence of a pulse (Figure 3, solid lines) there is no de novo DAG biosynthesis and the system is fixed in its basal state. In the basal state, both molecules reside in the cytosol with no possibility of translocation. In the presence of a pulse, the M/C ratios of both PKCα (Figure 3a, dashed line) and DGKζ (Figure 3b, dashed line) increase to their maximum levels followed by gradual clearance of signal. The temporal dynamics depicted in Figure 3 clearly show two phases of translocation. The first is an early phase in which the target and attenuator molecules of DAG signaling migrate from the cytosol to the plasma membrane. The first phase is followed by a second phase where both molecules translocate back to the cytosol. These results indicate that the migration of PKCα and DGKζ from the cytosol to the plasma membrane depends on local DAG concentration at the plasma membrane. My results also show the relative distribution of different forms of PKCα in different compartments (Figure 4). These results on the relative distribution of PKCα in different states describe that the biological function of the PKCα molecule in the proposed model is regulated through a four-step cycle. 11, 48 The four steps within this model are translocation, activation, redistribution/re-translocation, and deactivation. The model assumes that inactive but catalytically-competent PKCIα is stored in the cytosol. De novo synthesis generates a rather unstable naïve form of PKCIα which constitutively undergoes a sequence of ordered priming and autophosphorylations. This sequence produces a mature, inactive, phosphatase-resistant, and proteasome-resistant molecule.48 These phosphorylations are essential for PKCα stability and catalytic competence.48 The PKC life cycle is complex and modulated through precise and tightly-coupled molecular events.48 Evidence suggests that, before PKC becomes responsive to second messengers, it must first be phosphorylated at three conserved positions:48 Thr500, Thr641, and Ser660. This model does not attempt to model de novo synthesis or the subsequent processes of enzyme maturation through phosphorylation. This study assumes the cytosol contains sufficient amounts of mature, second messenger-responsive PKCIα enzyme. In these simulations, this is modeled by setting the initial conditions such that only the concentration of PKCIα is non-negligible. The concentration of all other forms of PKCα is initially set at negligibly small values.
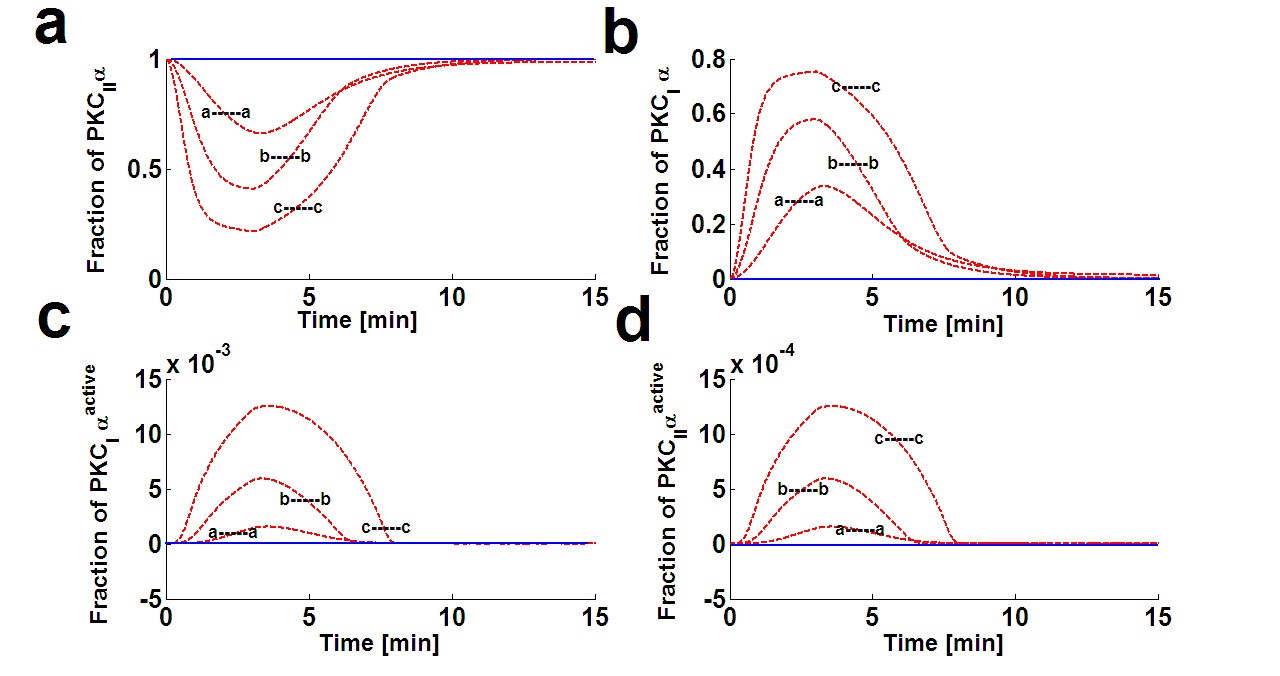
Figure 4. Dynamical characteristics of all four isoforms of PKCα. These temporal dynamics corresponds to results presented in figure 3 (relative distribution of PKCα and DGKζ in the membrane and cytosol compartments in response to Ang-II like stimulation). These results show that in the basal conditions all the PKCα resides in the dormant form in cytosol i.e., PKCIIα. However, on stimulation, the enzyme is distributed into all four forms i.e., PKCIIα, PKCIα, PKCIIαActive, and PKCIαActive. The extent and duration of this distribution is directly dependent on the Ang-II like stimulation and hence, on DAG. Here, three different levels of stimulation are used (Figure 3). The symbol a—a represents a pulse strength of 0.5, b—b represents a pulse strength of 2.0, and c—c represents the pulse strength of 6.0. Higher levels of the pulse (c—c) correspond to higher levels of DAG generation.
Experimental data indicate prolonged activation of PKCα in cardiomyocytes. The model proposed in this work is also calibrated against these results as shown in figure 5.
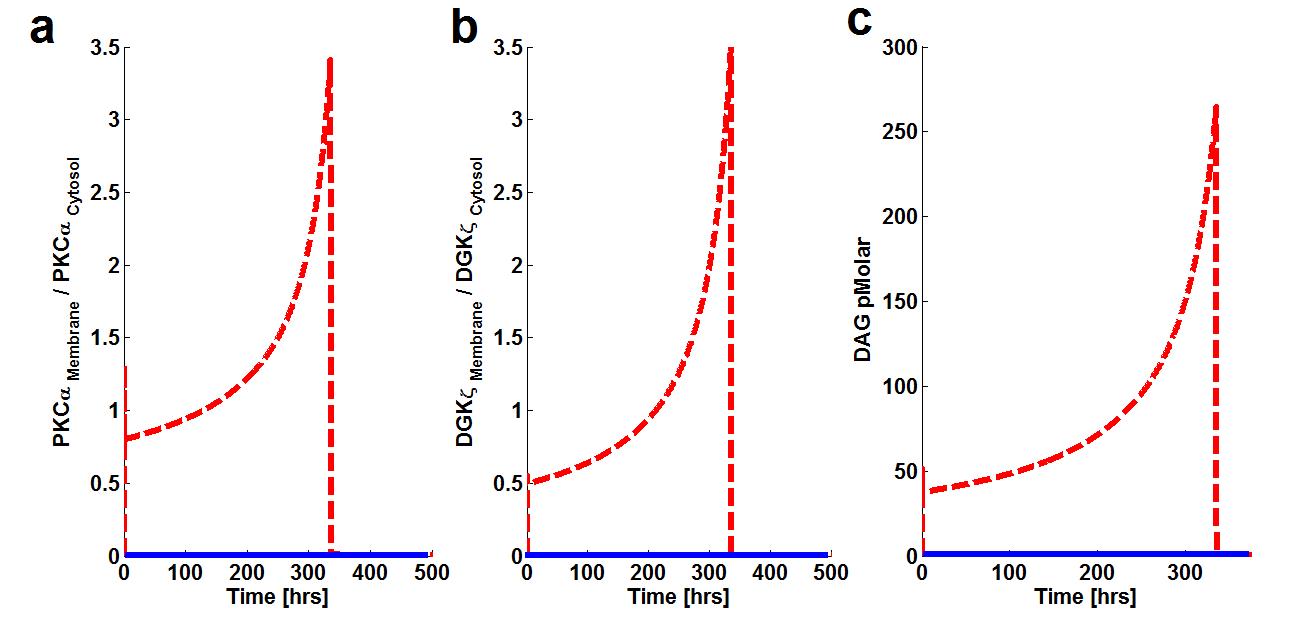
Figure 5. The effect of prolonged duration of stimulation on Membrane to cytosol (M/C) ratio of target & attenuator molecules of DAG signaling. These results show that prolonged pulse-like (duration of, 14 days) stimulation leads to the rapid generation of DAG and persistence. The generation of the second messenger, in turn, stimulates the translocation of both the target and attenuator molecules of DAG signaling from the cytosol to the membrane.
2.2 Effects of DGKζ Overexpression on the Translocation Characteristics of PKCα in Cardiomyocytes
The chance of survival after MI increases upon cardiac-specific overexpression of DGKζ. This is due to the attenuation of post-infarction LV remodeling. The survival rate almost doubles with DGKζ overexpression.14 Observations in transgenic mice indicate, that independent of hemodynamic effects, DGKζ overexpression attenuates angiotensin II-induced activation of DAG-PKCα signaling and subsequent contractile dysfunction.13-15Experimental observations show that, in wild type mouse hearts, angiotensin II induces the translocation of PKCα from the plasma membrane to the cytosol.13 This translocation event is blocked by DGKζ overexpression.13,15 I set out of address the question of why translocation is blocked by DGKζ overexpression by using a simple regulatory model for local DAG signaling. In this model’s simulations, DGKζ overexpression was implemented by adjusting the DGKζ-to-PKCα ratio under initial conditions. In the case where DGKζ is not overexpressed, the DGKζ to PKCα ratio is set at 1. For simulations mimicking two- and nine-fold overexpression over basal levels of DGKζ, this ratio is set at 2 and 9, respectively. Here, the effects of DGKζ overexpression on the migration of target and attenuator molecules of DAG signaling (Figures 6a and 6b and Figure 7) were tracked. Figure 6a shows that overexpression of DGKζ restricts PKCα translocation from the cytosol to the plasma membrane. At 2-fold DGKζ overexpression, a significant reduction in the M/C ratio of PKCα is observed. For 9-fold overexpression, a further reduction in the M/C ratio is observed and the M/C ratio for this case is less than one. At even higher levels of DGKζ overexpression, PKCα translocation to the plasma membrane is completely eliminated. These results also show that overexpression of DGKζ only slightly influences the migration characteristics of attenuator molecules of DAG signaling (Figure 6b). These results indicate that the maximum value of the M/C ratio of DGKζ decreases from 1.84 to 0.78 as the overexpression level increases from basal levels to 9-fold overexpression.
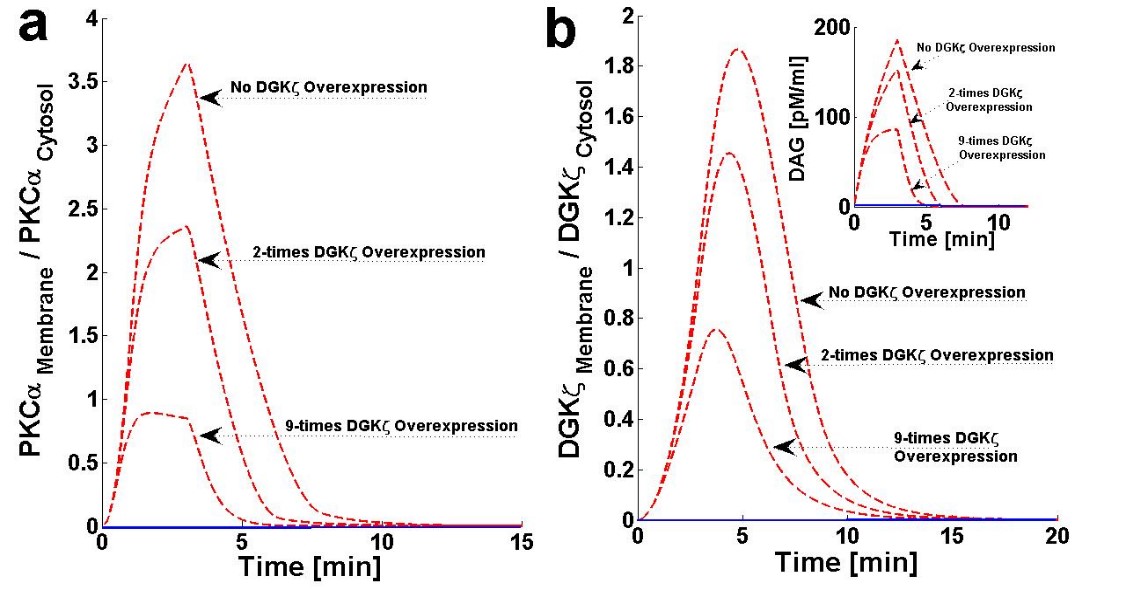
Figure 6. The effect of cardiac-specific overexpression of diacylglycerol kinase ζ (DGKζ) on the membrane to cytosol (M/C) ratio of target and attenuator molecules of DAG signaling in cardiomyocytes.
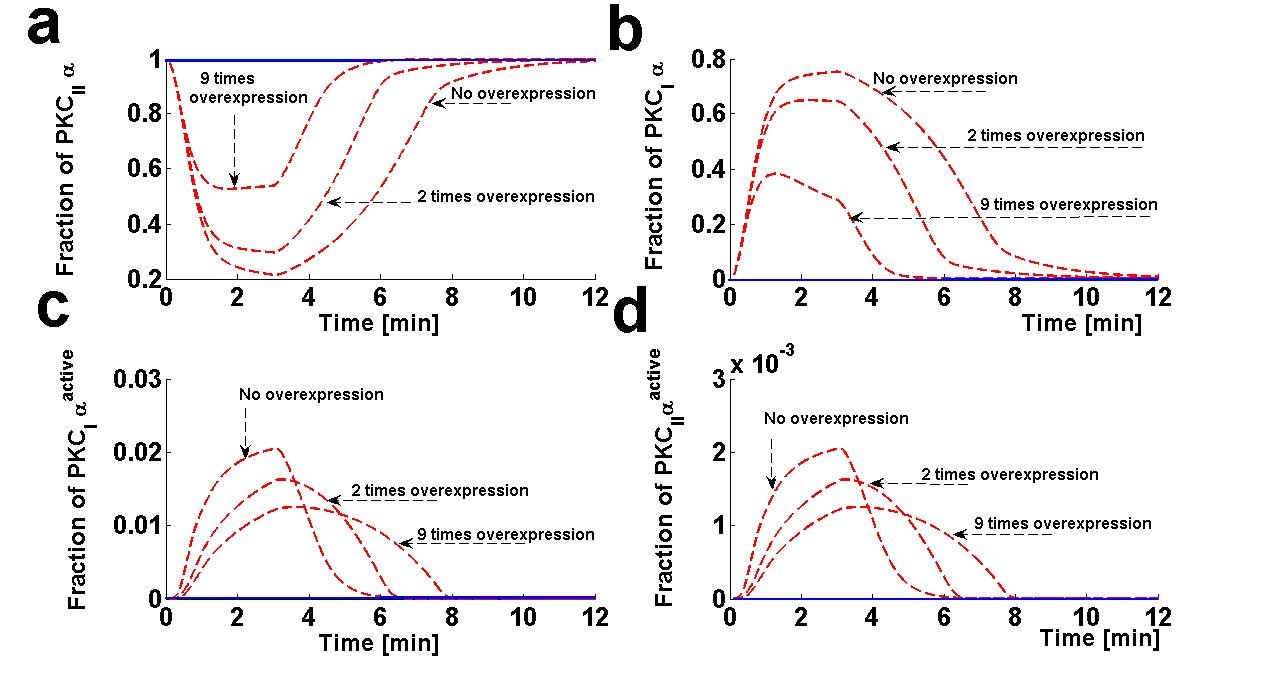
Figure 7. The effect of DGKζ overexpression on the dynamical characteristics of different forms of PKCα enzyme. These temporal dynamics correspond to results presented in figure 6 (relative distribution of PKCα and DGKζ in the membrane and cytosol compartments in response to the DGKζ overexpression at the pulse strength of 6.0).
In conclusion, a two-compartment model was developed for regulating DAG homeostasis in Ang II-induced heart failure. This computational model is a promising tool to study mechanisms of DAG regulation in the context of heart failure. This model may be used to identify novel therapeutic targets with the aim of improving survival and quality of life outcomes in heart failure patients.
References
- Ertl G, Gaudron P, Neubauer S, Bauer B, Horn M, Hu K, Tian R. Cardiac dysfunction and development of heart failure. Eur Heart J. 1993;14(Suppl A):33–37.
- Sun Y. Myocardial repair/remodelling following infarction: roles of local factors. Cardiovascular Res. 2009;Feb 15;81(3):482-90.
- Schwinger RH, Böhm M, Müller-Ehmsen J, Uhlmann R, Schmidt U, Stäblein A, Uberfuhr P, Kreuzer E, Reichart B, Eissner HJ. Effect of inotropic stimulation on the negative force-frequency relationship in the failing human heart. 1993;88:2267–2276.
- Dorn GW II, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. 2004;109: 150–158.
- Francis GS, Tang WH. Pathophysiology of congestive heart failure. Rev Cardiovascular Med. 2003;4:S14–S20.
- Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358.
- Colucci WS, Leatherman GF, Ludmer PL, et al-Adrenergic inotropic responsiveness of patients with heart failure: studies with intracoronary dobutamine infusion. Res. 1987;61(suppl I):I-82–I-86.
- Feldman AM, Bristow MR, Parmley WW, et al. Effects of vesnarinone on morbidity and mortality in patients with heart failure. Vesnarinone Study Group. N Engl J Med. 1993;329:149–155.
- Chatterjee K, De Marco T. Role of nonglycosidic inotropic agents: indications, ethics, and limitations. Med Clin North Am. 2003;87: 391– 418.
- Warner-Stevenson L. Clinical use of inotropic therapy for heart failure: looking backward or forward? Circulation. 2003;108:492– 497.
- Michael Hambleton, Harvey Hahn, Sven T. Pleger, Matthew C. Kuhn, Raisa Klevitsky, Andrew N. Carr, Thomas F. Kimball, Timothy E. Hewett, Gerald W. Dorn II, Walter J. Koch and Jeffery D. M. Pharmacological- and Gene Therapy-Based Inhibition of Protein Kinase Cα/β Enhances Cardiac Contractility and Attenuates Heart Failure. 2006;114:574-582.
- Hambleton M1, York A, Sargent MA, Kaiser RA, Lorenz JN, Robbins J, Molkentin JD. Inducible and myocyte-specific inhibition of PKCalpha enhances cardiac contractility and protects against infarction-induced heart failure. Am J Physiol Heart Circ Physiol. 2007 Dec;293(6):H3768-71.
- Steinberg, S.F. Cardiac Actions of Protein Kinase C Isoforms. 2012;27:130-139.
- Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW. Protein kinase C alpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res. 2003 Nov 28;93(11):1111-9.
- Arimoto T1, Takeishi Y, Takahashi H, Shishido T, Niizeki T, Koyama Y, Shiga R, Nozaki N, Nakajima O, Nishimaru K, Abe J, Endoh M, Walsh RA, Goto K, Kubota I. Cardiac-specific overexpression of diacylglycerol kinase zeta prevents Gq protein-coupled receptor agonist-induced cardiac hypertrophy in transgenic mice. 2006 Jan 3;113(1):60-6.
- Niizeki T1, Takeishi Y, Arimoto T, Takahashi H, Shishido T, Koyama Y, Goto K, Walsh RA, Kubota I. Cardiac-specific overexpression of diacylglycerol kinase zeta attenuates left ventricular remodeling and improves survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2007 Feb;292(2):H1105-12.
- Harada M1, Takeishi Y, Arimoto T, Niizeki T, Kitahara T, Goto K, Walsh RA, Kubota I. Diacylglycerol kinase zeta attenuates pressure overload-induced cardiac hypertrophy.Circ J. 2007 Feb;71(2):276-82.
- Niizeki T1, Takeishi Y, Kitahara T, Arimoto T, Ishino M, Bilim O, Suzuki S, Sasaki T, Nakajima O, Walsh RA, Goto K, Kubota I. Diacylglycerol kinase-epsilon restores cardiac dysfunction under chronic pressure overload: a new specific regulator of Galpha(q) signaling cascade. Am J Physiol Heart Circ Physiol. 2008 Jul;295(1):H245-55.
- Kehat I, Molkentin J. Molecular Pathways Underlying Cardiac Remodeling During Pathophysiological Stimulation. 2010; 122:2727-2735.
- Luo M, and Anderson M. E. Mechanisms of Altered Ca2+ Handling in Heart Failure. Res. 2013;113:690-708.
- Jarkko, P. Regulation of cardiac responses to increased load. Role of endothelin-1, angiotensin II and collagen XV. Department of Pharmacology and Toxicology and Biocenter Oulu, University of Oulu, P.O.Box 5000, FIN-90014 University of Oulu, Finland, Oulu, Finland 2002.
- Luo B1, Prescott SM, Topham MK. Association of diacylglycerol kinase zeta with protein kinase C alpha: spatial regulation of diacylglycerol signaling. J Cell Biol. 2003 Mar 17;160(6):929-37.
- Gharbi SI1, Rincón E, Avila-Flores A, Torres-Ayuso P, Almena M, Cobos MA, Albar JP, Mérida I. Diacylglycerol kinase ζ controls diacylglycerol metabolism at the immunological synapse. Biol. Cell. 2011 Nov;22(22):4406-14.
- Bai Luo, Debra S. Regier, Stephen M. Prescott, Matthew K. Topham. Diacylglycerol kinases. Cellular Signaling 16 (2004) 983-989.
- Takeishi Y1, Goto K, Kubota I. Role of diacylglycerol kinase in cellular regulatory processes: a new regulator for cardiomyocyte hypertrophy. Ther. 2007 Sep;115(3):352-9.
- Luo B, Prescott S.M, Topham M.K. Protein Kinase C{alpha} Phosphorylates and Negatively Regulates Diacylglycerol Kinase {zeta} J Biol Chem 2003 278:39542-39547.
- Gao T1, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283(10):6300-11.
- Warner-Stevenson L. Clinical use of inotropic therapy for heart failure: looking backward or forward? 2003;108:492– 497.