The implementation of cystic fibrosis (CF) transmembrane conductance regulator (CFTR) modulator drugs into clinical practice has been attaining remarkable therapeutic outcomes for CF, a life-threatening autosomal recessive genetic disease. However, there is elevated CFTR allelic heterogeneity, and various individuals carrying (ultra)rare CF genotypes remain without any approved modulator therapy. Novel translational model systems based on individuals’ own cells/tissue are now available and can be used to interrogate in vitro CFTR modulator responses and establish correlations of these assessments with clinical features, aiming to provide prediction of therapeutic effectiveness.
- airway cells
- bioassay
- biomarkers
- extracellular vesicles
- inflammation
- microRNA
- precision medicine
- organoids
- theratyping
1. Introduction
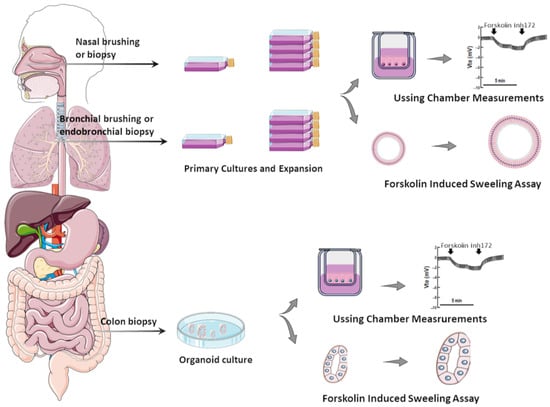
2. Laboratory Tools to Predict CFTR Modulator Effectiveness In Vivo
| Model System | CF Population/Genotypes | CFTR Modulator(s) | Key Findings/Correlations | Ref. |
|---|---|---|---|---|
| HNE cells | PwCF homozygous for p.Phe508del or carrying genotypes leading to minimal or residual CFTR function | LUMA TEZA IVA |
The level of CFTR rescue significantly correlated with the ppFEV1 change at 6 months in 8 PwCF treated with CFTR modulators. | [34] |
| Ten PwCF (<19 years) with a wide range of CFTR variants | LUMA IVA |
CFTR modulation produced changes in CFTR function in nasal and bronchial cell cultures. There was a correlation between the residual CFTR function in both cell types from PwCF with SCC between 60 and 90 mmol·L−1. |
[35] | |
| Five healthy subjects (they were not genetically tested) and eight PwCF with variants associated with minimal or residual CFTR function | – | CFTR-mediated transepithelial chloride currents measured in vitro through Ussing chamber assay correlated with the SCC of subjects. | [36] | |
| Twelve adults with CF with either the p.Gly551Asp or p.ArgR117His | IVA | A strong correlation between changes in SCC and in vitro CFTR activation was observed. Furthermore, a moderate correlation between in vitro CFTR activation and changes in ppFEV1 was reported. | [37] | |
| Seven PwCF (≤16 years, clinically stable) with residual CFTR function variants |
IVA | All subjects with decreased SCC in response to IVA treatment also had significant increases in chloride current in HNE cultures with IVA exposure. | [38] | |
| Healthy volunteers, PwCF with p.PheF508del/p.Arg117His-7T, p.PheF508del/p.PheF508del, p.PheF508del/p.Met284fs, p.Arg334Trp/c.406-1G>A, and p.Ser18ArgfsX16/p.Ser18ArgfsX16 | LUMA TEZA IVA |
In vitro CFTR chloride measurements correlated to changes in SCC. | [39] | |
| Five PwCF: one homozygous for p.Ser737Phe and four compound heterozygous for p.Ser737Phe | TEZA ELX IVA |
In vitro analysis demonstrated different levels of CFTR activity. Some degree of CFTR dysfunction was detected by evaluating chloride secretion in HNE cells derived from compound heterozygous subjects; CFTR activity was improved by IVA alone and even more by treatment with correctors. | [40] | |
| Eleven PwCF carrying FDA-eligible variants for ELX/TEZA/IVA and twenty-eight PwCF carrying non-eligible variants | TEZA ELX IVA |
There was a significant relationship between CFTR activity correction and changes in ppFEV1 or SCC. | [41] | |
| A 56-year-old male with CF and the p.Phe508del/p.Gln1291His genotype | TEZA ELX IVA |
In HNE cells, p.Phe508del/p.Gln1291His resulted in reduced baseline CFTR activity, and showed minimal response to ELX/TEZA/IVA (individually or in combination), aligning with the individual’s clinical evaluation as a non-responder to these drugs. | [42] | |
| Airway organoids | Nine healthy volunteers and three PwCF | LUMA IVA |
Organoids treated with modulators displayed similar effects to clinical response; LUMA/IVA treatment promoted greater responses in organoids from p.PheF508del-homozygous subjects. | [43] |
| Six healthy volunteers, nine PwCF homozygous for p.Phe508del, and ten with at least one non-p.Phe508del variant | LUMA IVA |
p.Phe508del/p.Phe508del organoids treated with LUMA/IVA exhibited a positive change in swelling responses; a relationship between organoid response to drug and in vivo clinical response was observed for three subjects treated. | [44] | |
| Eighteen PwCF and five healthy volunteers | TEZA ELX IVA |
In vitro responses to CFTR modulators correlated well with clinical measurements. | [45] | |
| Five PwCF: p.Phe508del/p.Phe508del, p.Phe508del/p.Gly542X, and F508del/G542X and p.Arg334Trp/p.Arg334Trp | LUMA IVA |
Responses of organoids correlated well with clinical findings. | [46] | |
| Intestinal organoids | Twelve healthy volunteers, four CF carriers (WT/p.Phe508del), thirty-five PwCF carrying class II and III variants, and eighteen PwCF carrying class IV and V variants |
LUMA IVA |
Responses of organoids to CFTR modulators correlated with outcome data from clinical trials. | [47] |
| Twenty-four PwCF: fifteen carrying at least one p.Ser1251Asn and nine carrying at least one (ultra)rare CFTR variant | LUMA IVA |
Responses to CFTR modulators in organoids correlated with in vivo measurements (SCC and ppFEV1). | [48] | |
| Thirty-four children with CF carrying a wide range of CFTR variants | – | Organoid swelling significantly correlated with SCC and ICM. | [49] | |
| Thirty-four PwCF with p.Phe508del/p.Phe508del | – | Variability in CFTR residual function appeared to contribute to the clinical heterogeneity and organoid swelling values; responses in organoids correlated with ppFEV1 and BMI. | [50] | |
| Ninety-seven PwCF with well-characterized and rare CFTR variants | LUMA IVA |
Measurements of residual CFTR function and rescue by CFTR modulators in organoids correlated with clinical data, namely changes in ppFEV1 and SCC. | [51] | |
| A 56-year-old female with CF carrying p.Phe508del/c.3717+5G>T | LUMA TEZA IVA |
No baseline swelling was observed in organoids, suggesting minimal CFTR function; however, limited swelling was detected after LUMA/IVA or TEZA/IVA treatment. | [52] | |
| Twenty-one PwCF homozygous for p.Phe508del | LUMA IVA |
No correlations were found between organoid swelling and changes in the in vivo biomarkers, namely SCC and NPD. | [53] | |
| A total of 173 PwCF carrying a wide range of CFTR variants | – | Organoid swelling values were associated with long-term ppFEV1 decline and the probability of developing different CF-related comorbidities, namely pancreatic insufficiency, CF-related liver disease, and CF-related diabetes. | [54] | |
| Fifteen PwCF homozygous for p.Phe508del, fifteen PwCF carrying p.Phe508del/class I variant, and twenty-two PwCF with rare variant non-eligible for CFTR modulator therapy | TEZA ELX IVA |
Responses to modulators in organoids from p.Phe508del/p.Phe508del or p.Phe508del/class I variant correlated with changes in ppFEV1. In CF organoids with 11 rare genotypes, CFTR function restoration was reported upon ELX/TEZA/IVA treatment. |
[55] | |
| A 19-year-old female with CF carrying p.Tyr515X/p.Arg334Trp | LUMA TEZA ELX IVA |
Organoids treated with IVA alone or in combination with correctors demonstrated similar rescue of CFTR-dependent fluid secretion. In vivo measurements demonstrated significant clinical improvements in SCC, ppFEV1, and respiratory symptoms after 7 days of IVA initiation that were sustained for the 9-month follow-up. |
[56] | |
| A 6-year-old male with CF carrying p.Phe508del/p.Glu217Gly-Gly509Asp | LUMA TEZA ELX IVA |
The p.Glu217Gly-Gly509Asp complex allele was characterized by a high residual function of the CFTR channel; organoid swelling values demonstrated rescue of CFTR function by all tested modulators. | [57] |
2.1. Primary Airway Cells Grown in Monolayers
2.2. Airway Organoids/Nasospheroids
2.3. Intestinal Organoids
This entry is adapted from the peer-reviewed paper 10.3390/jpm14010093
References
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662.
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462.
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065.
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073.
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080.
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723.
- Boon, M.; Verleden, S.E.; Bosch, B.; Lammertyn, E.J.; McDonough, J.E.; Mai, C.; Verschakelen, J.; Kemner-van de Corput, M.; Tiddens, H.A.W.; Proesmans, M.; et al. Morphometric Analysis of Explant Lungs in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 516–526.
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1.
- Sosnay, P.R.; White, T.B.; Farrell, P.M.; Ren, C.L.; Derichs, N.; Howenstine, M.S.; Nick, J.A.; De Boeck, K. Diagnosis of Cystic Fibrosis in Nonscreened Populations. J. Pediatr. 2017, 181S, S52–S57.e2.
- Cystic Fibrosis Mutation Database (CFTR1 Database). Available online: http://www.genet.sickkids.on.ca/ (accessed on 1 December 2023).
- Clinical and Function Translation of CFTR (CFTR2 Database). Available online: https://cftr2.org/ (accessed on 1 December 2023).
- Koch, C.; Cuppens, H.; Rainisio, M.; Madessani, U.; Harms, H.; Hodson, M.; Mastella, G.; Navarro, J.; Strandvik, B.; McKenzie, S.; et al. European Epidemiologic Registry of Cystic Fibrosis (ERCF): Comparison of major disease manifestations between patients with different classes of mutations. Pediatr. Pulmonol. 2001, 31, 1–12.
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38.
- Oliver, K.E.; Carlon, M.S.; Pedemonte, N.; Lopes-Pacheco, M. The revolution of personalized pharmacotherapies for cystic fibrosis: What does the future hold? Expert Opin. Pharmacother. 2023, 24, 1545–1565.
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34.
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913.
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672.
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231.
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035.
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023.
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948.
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819.
- Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410.
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321.
- Caputo, A.; Hinzpeter, A.; Caci, E.; Pedemonte, N.; Arous, N.; Di Duca, M.; Zegarra-Moran, O.; Fanen, P.; Galietta, L.J.V. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J. Pharmacol. Exp. Ther. 2009, 330, 786–791.
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245.
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748.
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2021, 14, 656–663.
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Cell Physiol. 2010, 298, C866–C874.
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885.
- Tomati, V.; Costa, S.; Capurro, V.; Pesce, E.; Pastorino, C.; Lena, M.; Sondo, E.; Di Duca, M.; Cresta, F.; Cristadoro, S.; et al. Rescue by elexacaftor-tezacaftor-ivacaftor of the G1244E cystic fibrosis mutation’s stability and gating defects are dependent on cell background. J. Cyst. Fibros. 2023, 22, 525–537.
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic fibrosis lung disease modifiers and their relevance in the new era of precision medicine. Genes 2021, 12, 562.
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021, 56, 1496–1503.
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 375.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3, e99385.
- Noel, S.; Servel, N.; Hatton, A.; Golec, A.; Rodrat, M.; Ng, D.R.S.; Li, H.; Pranke, I.; Hinzpeter, A.; Edelman, A.; et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl− currents in human nasal epithelial cells. J. Physiol. 2022, 600, 1515–1531.
- Debley, J.S.; Barrow, K.A.; Rich, L.M.; Singh, P.; McKone, E.F.; Nichols, D.P. Correlation between ivacaftor-induced CFTR activation in airway epithelial cells and improved lung function: A proof-of-concept study. Ann. Am. Thorac. Soc. 2020, 17, 1024–1027.
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479.
- Park, J.K.H.; Shrivastava, A.; Zhang, C.; Pollok, B.A.; Finkbeiner, W.E.; Gibb, E.R.; Ly, N.P.; Illek, B. Functional Profiling of CFTR-Directed Therapeutics Using Pediatric Patient-Derived Nasal Epithelial Cell Models. Front. Pediatr. 2020, 8, 536.
- Terlizzi, V.; Pesce, E.; Capurro, V.; Tomati, V.; Lena, M.; Pastorino, C.; Bocciardi, R.; Zara, F.; Centrone, C.; Taccetti, G.; et al. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators. Int. J. Mol. Sci. 2023, 24, 6576.
- Dreano, E.; Burgel, P.R.; Hatton, A.; Bouazza, N.; Chevalier, B.; Macey, J.; Leroy, S.; Durieu, I.; Weiss, L.; Grenet, D.; et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 2023, 62, 2300110.
- Allan, K.M.; Astore, M.A.; Kardia, E.; Wong, S.L.; Fawcett, L.K.; Bell, J.L.; Visser, S.; Chen, P.-C.; Griffith, R.; Jaffe, A.; et al. Q1291H-CFTR molecular dynamics simulations and ex vivo theratyping in nasal epithelial models and clinical response to elexacaftor/tezacaftor/ivacaftor in a Q1291H/F508del patient. Front. Mol. Biosci. 2023, 10, 1148501.
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2, e95734.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33.
- Anderson, J.D.; Liu, Z.; Odom, L.V.; Kersh, L.; Guimbellot, J.S. CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L119–L129.
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300.
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84.
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.
- de Winter-De Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529.
- de Winter-de Groot, K.M.; Berkers, G.; Marck-van der Wilt, R.E.P.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619.
- Ramalho, A.S.; Förstová, E.; Vonk, A.M.; Ferrante, M.; Verfailli, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekma, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426.
- Aalbers, B.L.; Brunsveld, J.E.; van der Ent, C.K.; van den Eijnden, J.C.; Beekman, J.M.; Heijerman, H.G.M. Forskolin induced swelling (FIS) assay in intestinal organoids to guide eligibility for compassionate use treatment in a CF patient with a rare genotype. J. Cyst. Fibros. 2022, 21, 254–257.
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of organoid swelling and in vivo biomarkers of cftr function to determine effects of lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for the f508del mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592.
- Muilwijk, D.; de Poel, E.; van Mourik, P.; Suen, S.W.F.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; Oppelaar, H.; Hagemeijer, M.C.; Berkers, G.; et al. Forskolin-induced Organoid Swelling is Associated with Long-term CF Disease Progression. Eur. Respir. J. 2022, 60, 2100508.
- Lefferts, J.W.; Bierlaagh, M.C.; Kroes, S.; Nieuwenhuijze, N.D.A.; Sonneveld van Kooten, H.N.; Niemöller, P.J.; Verburg, T.F.; Janssens, H.M.; Muilwijk, D.; van Beuningen, S.F.B.; et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. Int. J. Mol. Sci. 2023, 24, 14539.
- Mitropoulou, G.; Brandenberg, N.; Hoehnel, S.; Ceroni, C.; Balmpouzis, Z.; Blanchon, S.; Dorta, G.; Sauty, A.; Koutsokera, A. Rectal organoid-guided CFTR modulator therapy restores lung function in a CF patient with the rare 1677delTA/R334W genotype. Eur. Respir. J. 2022, 60, 2201341.
- Kondratyeva, E.; Melyanovskaya, Y.; Efremova, A.; Krasnova, M.; Mokrousova, D.; Bulatenko, N.; Petrova, N.; Polyakov, A.; Adyan, T.; Kovalskaia, V.; et al. Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele and Functional Evaluation of the CFTR Channel. Genes 2023, 14, 1705.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848.
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620.
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121.
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607.
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231.
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451.
- Müller, L.; Brighton, L.E.; Carson, J.L.; Fischer, W.A.; Jaspers, I. Culturing of human nasal epithelial cells at the air liquid interface. J. Vis. Exp. 2013, 80, e50646.
- Sette, G.; Lo Cicero, S.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur. Respir. J. 2021, 58, 2100908.
- Wu, X.; Wang, S.; Li, M.; Li, J.; Shen, J.; Zhao, Y.; Pang, J.; Wen, Q.; Chen, M.; Wei, B.; et al. Conditional reprogramming: Next generation cell culture. Acta Pharm. Sin. B 2020, 10, 1360–1381.
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324.
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381.
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289.
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568.
- Li, H.; Sheppard, D.N.; Hug, M.J. Transepithelial electrical measurements with the Ussing chamber. J. Cyst. Fibros. 2004, 3 (Suppl. S2), 123–126.
- Sheppard, D.N.; Gray, M.A.; Gong, X.; Sohma, Y.; Kogan, I.; Benos, D.J.; Scott-Ward, T.S.; Chen, J.-H.; Li, H.; Cai, Z.; et al. The patch-clamp and planar lipid bilayer techniques: Powerful and versatile tools to investigate the CFTR Cl− channel. J. Cyst. Fibros. 2004, 3, 101–108.
- Graeber, S.Y.; Balázs, A.; Ziegahn, N.; Rubil, T.; Vitzthum, C.; Piehler, L.; Drescher, M.; Seidel, K.; Rohrbach, A.; Röhmel, J.; et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 12365.
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might brushed nasal cells be a surrogate for CFTR modulator clinical response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126.
- Calucho, M.; Gartner, S.; Barranco, P.; Fernández-Álvarez, P.; Pérez, R.G.; Tizzano, E.F. Validation of nasospheroids to assay CFTR functionality and modulator responses in cystic fibrosis. Sci. Rep. 2021, 11, 15511.
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human nasal epithelial organoids for therapeutic development in cystic fibrosis. Genes 2020, 11, 603.
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945.
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265.
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772.
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289.
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal organoid morphology analysis (ROMA) as a promising diagnostic tool in cystic fibrosis. Thorax 2021, 76, 1146–1149.
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421.