Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Clostridioides difficile infection (CDI) is a leading nosocomial infection, posing a substantial public health challenge within the United States and globally. C. difficile releases toxins, which damage large intestinal epithelium, leading to toxic megacolon, sepsis, and even death.
- Clostridioides difficile infection
- microbiome-based therapy
- mucosal immunity
1. Introduction
1.1. Overview of Clostridioides difficile Infection (CDI)
The anaerobic gram-positive bacterium Clostridioides difficile, commonly referred to as C. diff, is one of the leading causes of antibiotic-associated diarrhea and infectious colitis in the world [1][2]. The organism commonly colonizes the gastrointestinal tracts of both healthy and chronically ill individuals of all age groups [3][4][5]. Disruption of the normal gut microbiota through administration of antibiotics can result in the proliferation of C. difficile with toxin production in capable strains resulting in CDI [4][6][7]. Most toxigenic strains of C. difficile produce toxin B (cytotoxin), with or without toxin A (enterotoxin) [7][8][9]. Some hypervirulent strains, such as Ribotype 027, produce a binary toxin (CDT), which rearranges the actin cytoskeleton of enterocytes, detrimentally affecting multiple cellular processes [1][7][8].
Accurate and timely diagnosis of CDI is an important yet challenging issue facing healthcare systems today [3][4]. CDI is a common misdiagnosis for many patients, due to the high frequency of C. difficile colonization, varying individual tolerances to C. difficile toxins, numerous causes of diarrhea besides C. difficile, and common pitfalls of routine diagnostic testing [3][4][5][6][10]. Prompt initiation of appropriate treatment is needed to prevent the development of severe manifestations of CDI (e.g., pseudomembranous colitis, toxic megacolon, ileus, septic shock); however, unnecessary treatment can lead to the establishment of multidrug-resistant organisms in patients’ colonic flora [3][4][5][6]. Diagnostic stewardship, utilizing astute clinical judgment and judicious use of microbiological testing, is key to maximizing diagnostic accuracy [3][4][5][6].
Given the challenges associated with accurately diagnosing CDI, recent advancements in clinical tests and therapies have become increasingly significant in improving patient outcomes. According to a review of the diagnosis and management of CDI in adults, new clinical tests and therapies have become available, and clinical practice guidelines were updated [11]. The Infectious Diseases Society of America (IDSA) and the Society for Healthcare Epidemiology of America (SHEA) have published a clinical practice guideline on the management of CDI in adults [12].
1.2. Public Health Significance of CDI
With just over 200,000 cases annually resulting in ~12,000 deaths, CDI is a major public health concern in the U.S [13]. The Centers for Disease Control and Prevention (CDC) lists C. difficile as an urgent threat requiring aggressive preventative measures [13]. Reducing the amounts of CDI cases by optimizing modifiable risk factors (e.g., unnecessary antibiotic exposure, hospitalization prolongation) not only reduces the associated morbidity and mortality of CDI but also leads to a substantial reduction in the substantial healthcare costs associated with this infection [4]. It is estimated that CDI costs USD 1 billion in associated U.S. healthcare spending, based on the most recent CDC statistics [13].
Stringent infection prevention and control practices are required to prevent the spread of this spore-forming bacterium in healthcare settings [3][4]. Recommended infection control practices include isolation of patients with CDI, wearing gloves and gowns with disposable equipment use during patient encounters, hand washing with soap and water after patient contact, and use of sporicidal cleaning agents [3][4]. Antibiotic stewardship programs are effective in reducing CDI cases and should be established in most healthcare systems [3][4]. Reducing unnecessary antibiotic administration and utilizing antimicrobial agents with lower association with CDI should be the goal of these programs [4].
1.3. The Challenge of CDI Recurrence
One of the most challenging issues facing the management of CDI is its propensity for recurrence, commonly resulting in multiple subsequent episodes [3][12][14]. Not only does this contribute to this disease process’s morbidity and mortality, but it also leads to an exponential rise in healthcare-associated costs [12][15]. Disruption of the gut microbiota leading to the development of CDI and following treatment of it predisposes individuals to recurrence [16]. The inability of the host’s immune system to fully clear the organism from the gastrointestinal tract is also a major factor in recurrence [16].
Several new treatment options have shown promise in reducing the frequency of CDI recurrence [3][12][17]. Vancomycin and metronidazole have been the two agents most commonly used for the treatment of CDI; however, both demonstrate antibacterial effects against beneficial bacteria in the normal gut flora, leading to persistent dysbiosis [3][12][18]. Fidaxomicin has become the preferred treatment agent in non-fulminant CDI cases, due to its narrow-spectrum antibacterial activity leading to the preservation of the gut microbiota and less CDI recurrences [3][12][18][19]. The MODIFY I and MODIFY II clinical trials showed that bezlotoxumab, a monoclonal antibody targeting the C. difficile toxin B, is effective in reducing recurrence following primary and recurrent CDI when administered with standard-of-care antibiotics [20]. Bezlotoxumab administration should be considered in patients with recurrence or at high-risk for recurrence [3][12][18]. Fecal microbiota transplantation (FMT) is a novel option for individuals with multiple CDI recurrences despite appropriate antibiotic therapy [3][12][18][21][22]. Stool from donors with healthy gut microbiota are transplanted via colonoscopy or oral capsule to restore a functional gut flora [23]. FMT has been shown to be highly successful in preventing CDI recurrence, but the transplantation of multi-drug resistant organisms and toxin-producing bacteria have been reported [21][22][24][25][26][27]. Although antibiotics are the primary treatment for initial CDI, their effectiveness is only partial. Additionally, the use of antibiotics can lead to persistent dysbiosis, contributing to recurrent infections in a significant number of patients [14][28][29]. Recent studies highlight the role of immune responses and cytokines released during acute CDI in the disease’s pathogenesis. Specifically, the modulation of cytokines has been proven to influence CDI outcomes. For instance, elevating the levels of certain cytokines, such as IL33 and IL25, enhances host protection [30][31]. Mice treated with these cytokines exhibited less mucosal damage and greater resistance against CDI compared to the untreated group. Furthermore, depleting immune cells has been shown to impact the outcome of CDI pathogenesis [32][33]. These findings underscore the potential of host immune-based therapy in managing CDI.
2. Host Immune Response to C. difficile Infection
2.1. Innate Immune Response
The innate defense mechanisms against C. difficile infection include the endogenous microbial flora, the mucus barrier, intestinal epithelial cells, and the mucosal immune cells.
C. difficile primarily produces toxin A and toxin B (with certain strains also producing binary toxins), causing disruption to the intestinal epithelium, leading to activation of the immune responses in the lamina propria of the colon. These toxins have a profound impact on innate immune defenses, triggering the release of various proinflammatory mediators, such as cytokines and chemokines, promoting the recruitment and activation of diverse innate immune cells (Figure 1). Further, the disrupted mucosal barrier, due to toxin-mediated damage, allows commensal bacteria to translocate into the lamina propria and systemic circulation, leading to robust inflammatory responses.
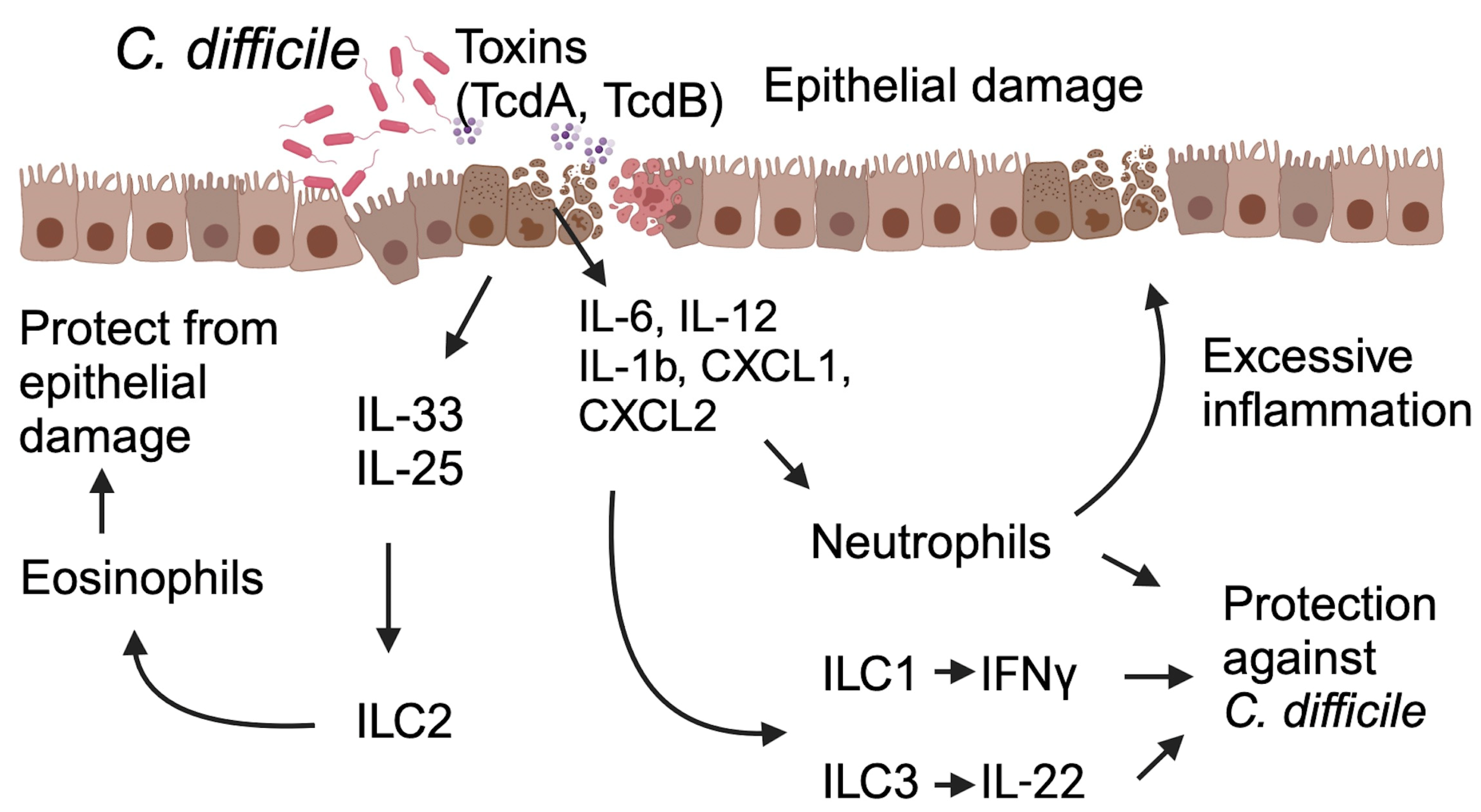
Figure 1. This schematic illustrates the host’s immune response during C. difficile infection and its impact on the infection’s outcomes. C. difficile toxins A and B lead to epithelial damage, triggering immune-cell activation and the release of cytokines and chemokines from both immune cells and the damaged epithelium. These signaling molecules, in turn, activate innate lymphoid cells (ILCs) and promote the recruitment of neutrophils to the site of injury. Activated ILC1 and ILC3 release Interferon IFNγ and IL-22, conferring protection against C. difficile. Neutrophils, though known for their protective function, may also have the potential to cause epithelial damage and have a detrimental impact on the outcome of CDI. Specific cytokines, such as IL-25 and IL-33, induce a type 2 immune response, enhancing host defense by increasing eosinophil infiltration and activation at the site of damage. Abbreviations: ILC1 (Innate Lymphoid Cell 1), ILC2 (Innate Lymphoid Cell 2), ILC3 (Innate Lymphoid Cell 3), TcdA (C. difficile Toxin A), TcdB (C. difficile Toxin B).
In response to C. difficile toxins and their associated damage, intestinal epithelial cells, and innate immune cells in the lamina propria release proinflammatory cytokines (IFNγ, IL-12, IL-6, IL-23, IL-1β, etc.) and chemokines (CXCL1, CXCL2, and CXCL5), leading to the recruitment of neutrophils to the site of infection [34][35][36][37].
Proteins involved in the signaling of the innate immune response, such as nucleotide-binding and oligomerization domain 1 (NOD1), myeloid differentiation factor 88 (MyD88), and adaptor protein for inflammasome, known as apoptosis-associated speck-like protein containing a CARD (ASC), play a role in C. difficile pathogenesis. Studies show that mice deficient in Nod1, MyD88, and ASC signaling have decreased levels of CXCL1 and infiltrating neutrophils in their colons, which is associated with enhanced disease severity and mortality, compared to wild-type mice [32][38][39]. It is worth mentioning that the role of neutrophils and their effect on host factors in CDI susceptibility is complex and context-dependent, with various pathways and cell types involved in the overall pathogenesis. For example, Jarchum et al. found that depletion of neutrophils in mice through antibody-mediated Gr1+ (Ly6G) depletion increases the disease severity and mortality during CDI [32]. This finding is supported by the clinical observation that in hospitalized leukemia and allogeneic hematopoietic stem cell transplant patients, neutropenia is positively correlated with CDI susceptibility and recurrent CDI [40][41]. In contrast, another study found that blocking of neutrophil infiltration in the colon by anti-CD18 (leukocyte adhesion molecule) treatment in rabbit leads to decreased Toxin A-mediated enterotoxicity, compared to non-anti-CD18 treated mice [42]. Similarly, another study in a mouse model of CDI showed that mice treated with macrophage migration inhibitory factor (MIF) blocking antibody demonstrated improved disease severity and survival, which is associated with reduced neutrophil recruitment in the colon [43]. However, a recent study by Chen et al. found no differences in CDI severity when they depleted neutrophils using anti-Ly6G antibodies [33]. The differing roles of neutrophils in CDI outcomes in various studies may result from variances in animal models, C. difficile strains, experimental design, and host-specific factors. Other innate immune cells, such as eosinophils, play a protective role in CDI. In studies using mice, Buonomo et al. show that mice adoptive transferred of eosinophils are more protected against CDI [30], supported by a clinical study showing that, in patients, the eosinopenia in peripheral blood at the time of CDI diagnosis was associated with higher mortality [44].
C. difficile and its toxins can activate surface and intracellular innate immune sensors, including TLR4, TLR5, and inflammasome signaling pathways [38][45][46][47]. Depletion of TLR4-signaling pathways in mice results in an increased bacterial burden and disease severity during CDI [45]. On the other hand, TLR5 deficiency did not affect survival, but stimulating TLR5 with flagellin provided protection against CDI, likely due to its positive impact on the intestinal epithelial layer [46].
In vitro, infection of murine peritoneal macrophages with a toxigenic strain of C. difficile results in the release of the proinflammatory cytokine pro-IL-1β, a process that is dependent on MyD88 and, to some extent, TLR2 [48]. In the same study, toxigenic C. difficile activated inflammasome through the ATP-P2X7 pathway, leading to caspase-1-dependent pyroptosis [48]. In a different study, stimulating the J774A.1 murine macrophage cell line with C. difficile’s surface layer protein (SLP) led to increased expression of TLR2, TLR4, and MHCII [49]. Furthermore, the activation of TLR4 using SLPs triggered p38 signaling, leading to increased production of IL-1β, IL-6, TNF-α, IL-12p40, and the upregulation of chemokines like MIP-1α, MIP-2, and MCP [49].
Recent studies have shed light on the protective role of innate lymphoid cells (ILCs) in acute CDI. Unlike adaptive immune cells like T and B cells, ILCs, which include ILC1, ILC2, and ILC3, lack antigen-specific receptors [50]. However, they possess the ability to detect cytokines and chemokines and are pivotal in orchestrating immune responses against infections and maintaining tissue homeostasis [50]. During CDI, when commensal microorganisms, pathobiont bacteria, or C. difficile toxins translocate across the gut barrier into deeper tissues, this triggers the activation of ILCs. A study showed that Nfil3-deficient (Nfil3-/-) mice, which exhibit defects in the development and functionality of ILCs, demonstrated heightened susceptibility to acute CDI compared to their wild-type counterparts [51]. Nfil3-/- mice infected with C. difficile manifest increased weight loss and higher mortality rates than their wild-type (WT) counterparts. Another study conducted by Abt and colleagues uncovered the protective function of IFNγ-expressing ILC1 as part of the host’s defense against CDI, complemented by the role of IL22-expressing ILC3 [52]. Additionally, another research group has demonstrated the role of ILC2s in protection against C. difficile colitis. For instance, Frisbee et al. showed that ILC2 provides defense against acute CDI, especially when activated by IL-33 [31]. Notably, treating C. difficile-infected mice with IL-33 leads to reduced neutrophil levels and increased eosinophils in the colon, resulting in type 2-associated mucosal immunity. Similarly, another study further underscored the IL-25-mediated role of ILC2s in defending against CDI [30]. In parallel, other researchers also demonstrated the protective role of ILC3 in safeguarding against CDI, primarily through the interleukin-22-mediated effect [53][54].
The aforementioned studies indicated the crucial role of innate immunity in the initial containment of infections and its contribution to resolving CDI. Nonetheless, the recurrence of episodes implies that adaptive immune cells may be fundamentally involved. This is attributed to their capacity to respond in an antigen-specific manner and generate memory, thereby providing additional protection.
2.2. Adaptive Immune Response
The defining feature of the adaptive immune system is the clonal expansion of lymphocytes, resulting in a durable and highly specific response. Typically, memory T and B cells orchestrate more robust and rapid immune reactions to pathogens—rapidly multiplying, generating effector cytokines, and executing various effector functions [55]. However, an impaired immune response to pathogens can result in recurring infections.
Around 20 to 35% of individuals treated for CDI encounter at least one additional episode within 2 to 8 weeks of their initial CDI treatment [17][41]. These subsequent events can manifest as either a relapse with the same C. difficile strain or as a reinfection with a different strain [56]. One possible factor contributing to susceptibility to recurrent CDI is the ongoing disturbance of gut microbiota diversity [14][57]. This, in conjunction with a weakened host response, may play a role, since low levels of serum antibodies against toxins A and B have been linked to the CDI recurrences [58][59].
In CDI, immunoglobulins such as systemic IgG and mucosal IgA significantly influence disease outcomes. In human CDI cases, the disease’s severity shows an inverse correlation with the levels of toxin-specific IgA and IgG antibodies in the serum and secretory intestinal IgA [59][60]. Furthermore, in fecal samples, individuals experiencing a single CDI episode demonstrated significantly higher anti-TcdA IgA titers, compared to those with recurrent CDI [61]. Studies also have found an inverse relationship between IgG levels against toxin B and disease severity, as well as CDI recurrence. Notably, patients possessing antibodies against toxin B exhibit enhanced protection compared to those with antibodies against toxin A [62]. Further, monoclonal antibodies targeting TcdB have demonstrated greater efficacy in CDI treatment [20], indicating TcdB’s higher toxicity compared to TcdA. It is important to note that while elevated antibodies against C. difficile toxins can guard against the disease and its relapse, they do not prevent C. difficile colonization in the colon.
Most of the research pertaining to the host’s adaptive immunity in CDI has primarily concentrated on humoral responses. On the other hand, the importance of T follicular helper (Tfh) cells, which play a critical role in generating plasma cells that produce antibodies and long-lasting memory B cells, has only recently begun to be investigated. The exploration of various subsets of T cells and their effector functions against C. difficile is still in its early stages.
At the germinal center, Tfh cells play a crucial role in aiding B cells, ultimately leading to the differentiation of activated B cells into plasma cells and memory B cells [63][64]. Consequently, Tfh cells are instrumental in conferring antibody-mediated protection to the intestinal mucosa against pathogens. In a murine model of recurrent C. difficile infection, Amani et al. observed a notable expansion in the population of lymph node resident Tfh cells, encompassing both germinal center and non-germinal center Tfh cells, following CDI when compared to uninfected mice [65]. Despite the observed expansion of Tfh cells, the B cell response proved insufficient in preventing disease recurrence. Furthermore, the authors demonstrated that CDI failed to elicit a robust B cell memory response.
Studies have demonstrated that C. difficile strains trigger a CD4+ T cell response. For example, the hypervirulent C. difficile R20291 strain elicits a robust Th1 and Th17 response, as evidenced by an increased presence of IFNγ+ and IL-17A+ CD4+ T cells, when compared to the non-virulent C. difficile 630 strain in co-culture with murine splenocyte and bone-marrow-derived dendritic cells [66]. In a clinical study, C. difficile-infected patients exhibited a shift in immune responses from Th1 to Th17 or Th2 as disease severity increased [67]. Studies by other investigators also demonstrated the role of T cells in CDI. Notably, young children display resistance to CDI, and a study in mice has highlighted the role of IL-17A produced by γδ T cells in this resistance [33]. Neonatal mice, resistant to CDI, demonstrated substantial IL-17 production by RORγt+ γδ T cells [33]. However, this protective effect was lost upon depletion of these IL-17-producing T cells.
Regulatory T cells (Treg), a subset of CD4+ T cells, are crucial for maintaining intestinal immune tolerance and homeostasis. Their specific impact on a host’s susceptibility to acute CDI and relapse remains unclear. However, a recent study highlighted the vital role of Treg cells in successfully engrafting fecal microbiota and clearing chronic CDI in mice [68]. Mechanistically, depleting Treg cells causes an exaggerated immune response in the colon, hindering C. difficile clearance by impeding the engraftment of microbial populations derived from fecal microbiota transplantation (FMT).
This entry is adapted from the peer-reviewed paper 10.3390/tropicalmed8120506
References
- Liu, C.; Monaghan, T.; Yadegar, A.; Louie, T.; Kao, D. Insights into the Evolving Epidemiology of Clostridioides Difficile Infection and Treatment: A Global Perspective. Antibiotics 2023, 12, 1141.
- Ikuta, K.S.; Swetschinski, L.R.; Aguilar, G.R.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Weaver, N.D.; Wool, E.E.; Han, C.; Hayoon, A.G.; et al. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248.
- Kelly, C.R.; Fischer, M.; Allegretti, J.R.; LaPlante, K.; Stewart, D.B.; Limketkai, B.N.; Stollman, N.H. ACG Clinical Guidelines: Prevention, Diagnosis, and Treatment of Clostridioides Difficile Infections. Am. J. Gastroenterol. 2021, 116, 1124–1147.
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium Difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, e1–e48.
- Cotter, J.M.; Thomas, J.; Birkholz, M.; Brittan, M.; Ambroggio, L.; Dolan, S.; Pearce, K.; Todd, J.; Dominguez, S.R. Impact of Multiplex Testing on the Identification of Pediatric Clostridiodes Difficile. J. Pediatr. 2020, 218, 157–165.
- Rock, C.; Maragakis, L.L. Diagnostic Stewardship for Clostridiodes Difficile Testing: From Laxatives to Diarrhea and Beyond. Clin. Infect. Dis. 2020, 71, 1479–1480.
- Cowardin, C.A.; Buonomo, E.L.; Saleh, M.M.; Wilson, M.G.; Burgess, S.L.; Kuehne, S.A.; Schwan, C.; Eichhoff, A.M.; Koch-Nolte, F.; Lyras, D.; et al. The Binary Toxin CDT Enhances Clostridium Difficile Virulence by Suppressing Protective Colonic Eosinophilia. Nat. Microbiol. 2016, 1, 16108.
- Jeon, C.-H.; Kim, S.-H.; Wi, Y.M. Prevalence of Non-Toxigenic Clostridioides Difficile in Diarrhoea Patients and Their Clinical Characteristics. Antibiotics 2023, 12, 1360.
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The Role of Toxin A and Toxin B in Clostridium Difficile Infection. Nature 2010, 467, 711–713.
- Pollock, N.R.; Banz, A.; Chen, X.; Williams, D.; Xu, H.; Cuddemi, C.A.; Cui, A.X.; Perrotta, M.; Alhassan, E.; Riou, B.; et al. Comparison of Clostridioides Difficile Stool Toxin Concentrations in Adults With Symptomatic Infection and Asymptomatic Carriage Using an Ultrasensitive Quantitative Immunoassay. Clin. Infect. Dis. 2019, 68, 78–86.
- Rao, K.; Malani, P.N. Diagnosis and Treatment of Clostridioides (Clostridium) Difficile Infection in Adults in 2020. JAMA 2020, 323, 1403–1404.
- Johnson, S.; Lavergne, V.; Skinner, A.M.; Gonzales-Luna, A.J.; Garey, K.W.; Kelly, C.P.; Wilcox, M.H. Clinical Practice Guideline by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA): 2021 Focused Update Guidelines on Management of Clostridioides Difficile Infection in Adults. Clin. Infect. Dis. 2021, 73, 755–757.
- The Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 30 October 2023).
- Zanella Terrier, M.C.; Simonet, M.L.; Bichard, P.; Frossard, J.L. Recurrent Clostridium Difficile Infections: The Importance of the Intestinal Microbiota. World J. Gastroenterol. 2014, 20, 7416–7423.
- Guh, A.Y.; Mu, Y.; Winston, L.G.; Johnston, H.; Olson, D.; Farley, M.M.; Wilson, L.E.; Holzbauer, S.M.; Phipps, E.C.; Dumyati, G.K.; et al. Trends in U.S. Burden of Clostridioides Difficile Infection and Outcomes. N. Engl. J. Med. 2020, 382, 1320–1330.
- Petrosillo, N. Tackling the Recurrence of Clostridium Difficile Infection. Med. Mal. Infect. 2018, 48, 18–22.
- Hui, W.; Li, T.; Liu, W.; Zhou, C.; Gao, F. Fecal Microbiota Transplantation for Treatment of Recurrent C. Difficile Infection: An Updated Randomized Controlled Trial Meta-Analysis. PLoS ONE 2019, 14, e0210016.
- van Prehn, J.; Reigadas, E.; Vogelzang, E.H.; Bouza, E.; Hristea, A.; Guery, B.; Krutova, M.; Norén, T.; Allerberger, F.; Coia, J.E.; et al. European Society of Clinical Microbiology and Infectious Diseases: 2021 Update on the Treatment Guidance Document for Clostridioides Difficile Infection in Adults. Clin. Microbiol. Infect. 2021, 27 (Suppl. 2), S1–S21.
- Chen, J.; Gong, C.L.; Hitchcock, M.M.; Holubar, M.; Deresinski, S.; Hay, J.W. Cost-Effectiveness of Bezlotoxumab and Fidaxomicin for Initial Clostridioides Difficile Infection. Clin. Microbiol. Infect. 2021, 27, 1448–1454.
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C.; et al. Bezlotoxumab for Prevention of Recurrent Clostridium Difficile Infection. N. Engl. J. Med. 2017, 376, 305–317.
- Madoff, S.E.; Urquiaga, M.; Alonso, C.D.; Kelly, C.P. Prevention of Recurrent Clostridioides Difficile Infection: A Systematic Review of Randomized Controlled Trials. Anaerobe 2020, 61, 102098.
- Kelly, C.R.; Khoruts, A.; Staley, C.; Sadowsky, M.J.; Abd, M.; Alani, M.; Bakow, B.; Curran, P.; McKenney, J.; Tisch, A.; et al. Effect of Fecal Microbiota Transplantation on Recurrence in Multiply Recurrent Clostridium Difficile Infection: A Randomized Trial. Ann. Intern. Med. 2016, 165, 609–616.
- Kao, D.; Roach, B.; Silva, M.; Beck, P.; Rioux, K.; Kaplan, G.G.; Chang, H.-J.; Coward, S.; Goodman, K.J.; Xu, H.; et al. Effect of Oral Capsule- vs Colonoscopy-Delivered Fecal Microbiota Transplantation on Recurrent Clostridium Difficile Infection: A Randomized Clinical Trial. JAMA 2017, 318, 1985–1993.
- Khanna, S.; Kraft, C.S. Fecal Microbiota Transplantation: Tales of Caution. Clin. Infect. Dis. 2021, 72, e881–e882.
- Zellmer, C.; Sater, M.R.A.; Huntley, M.H.; Osman, M.; Olesen, S.W.; Ramakrishna, B. Shiga Toxin-Producing Escherichia Coli Transmission via Fecal Microbiota Transplant. Clin. Infect. Dis. 2021, 72, e876–e880.
- Minkoff, N.Z.; Aslam, S.; Medina, M.; Tanner-Smith, E.E.; Zackular, J.P.; Acra, S.; Nicholson, M.R.; Imdad, A. Fecal Microbiota Transplantation for the Treatment of Recurrent Clostridioides Difficile (Clostridium Difficile). Cochrane Database Syst. Rev. 2023, 4, CD013871.
- Kelly, C.R.; Yen, E.F.; Grinspan, A.M.; Kahn, S.A.; Atreja, A.; Lewis, J.D.; Moore, T.A.; Rubin, D.T.; Kim, A.M.; Serra, S.; et al. Fecal Microbiota Transplantation Is Highly Effective in Real-World Practice: Initial Results from the FMT National Registry. Gastroenterology 2021, 160, 183–192.e3.
- Abujamel, T.; Cadnum, J.L.; Jury, L.A.; Sunkesula, V.C.K.; Kundrapu, S.; Jump, R.L.; Stintzi, A.C.; Donskey, C.J. Defining the Vulnerable Period for Re-Establishment of Clostridium Difficile Colonization after Treatment of C. Difficile Infection with Oral Vancomycin or Metronidazole. PLoS ONE 2013, 8, e76269.
- Lewis, B.B.; Buffie, C.G.; Carter, R.A.; Leiner, I.; Toussaint, N.C.; Miller, L.C.; Gobourne, A.; Ling, L.; Pamer, E.G. Loss of Microbiota-Mediated Colonization Resistance to Clostridium Difficile Infection with Oral Vancomycin Compared With Metronidazole. J. Infect. Dis. 2015, 212, 1656–1665.
- Buonomo, E.L.; Cowardin, C.A.; Wilson, M.G.; Saleh, M.M.; Pramoonjago, P.; Petri, W.A.J. Microbiota-Regulated IL-25 Increases Eosinophil Number to Provide Protection during Clostridium Difficile Infection. Cell Rep. 2016, 16, 432–443.
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 Drives Group 2 Innate Lymphoid Cell-Mediated Protection during Clostridium Difficile Infection. Nat. Commun. 2019, 10, 2712.
- Jarchum, I.; Liu, M.; Shi, C.; Equinda, M.; Pamer, E.G. Critical Role for MyD88-Mediated Neutrophil Recruitment during Clostridium Difficile Colitis. Infect. Immun. 2012, 80, 2989–2996.
- Chen, Y.-S.; Chen, I.-B.; Pham, G.; Shao, T.-Y.; Bangar, H.; Way, S.S.; Haslam, D.B. IL-17-Producing Γδ T Cells Protect against Clostridium Difficile Infection. J. Clin. Investig. 2020, 130, 2377–2390.
- McDermott, A.J.; Falkowski, N.R.; McDonald, R.A.; Pandit, C.R.; Young, V.B.; Huffnagle, G.B. Interleukin-23 (IL-23), Independent of IL-17 and IL-22, Drives Neutrophil Recruitment and Innate Inflammation during Clostridium Difficile Colitis in Mice. Immunology 2016, 147, 114–124.
- Warny, M.; Keates, A.C.; Keates, S.; Castagliuolo, I.; Zacks, J.K.; Aboudola, S.; Qamar, A.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. P38 MAP Kinase Activation by Clostridium Difficile Toxin A Mediates Monocyte Necrosis, IL-8 Production, and Enteritis. J. Clin. Investig. 2000, 105, 1147–1156.
- Kim, J.M.; Lee, J.Y.; Yoon, Y.M.; Oh, Y.-K.; Youn, J.; Kim, Y.-J. NF-Kappa B Activation Pathway Is Essential for the Chemokine Expression in Intestinal Epithelial Cells Stimulated with Clostridium Difficile Toxin A. Scand. J. Immunol. 2006, 63, 453–460.
- McDermott, A.J.; Frank, C.R.; Falkowski, N.R.; McDonald, R.A.; Young, V.B.; Huffnagle, G.B. Role of GM-CSF in the Inflammatory Cytokine Network That Regulates Neutrophil Influx into the Colonic Mucosa during Clostridium Difficile Infection in Mice. Gut Microbes 2014, 5, 476–484.
- Hasegawa, M.; Yamazaki, T.; Kamada, N.; Tawaratsumida, K.; Kim, Y.-G.; Núñez, G.; Inohara, N. Nucleotide-Binding Oligomerization Domain 1 Mediates Recognition of Clostridium Difficile and Induces Neutrophil Recruitment and Protection against the Pathogen. J. Immunol. 2011, 186, 4872–4880.
- Hasegawa, M.; Kamada, N.; Jiao, Y.; Liu, M.Z.; Núñez, G.; Inohara, N. Protective Role of Commensals against Clostridium Difficile Infection via an IL-1β-Mediated Positive-Feedback Loop. J. Immunol. 2012, 189, 3085–3091.
- Luo, R.; Greenberg, A.; Stone, C.D. Outcomes of Clostridium Difficile Infection in Hospitalized Leukemia Patients: A Nationwide Analysis. Infect. Control Hosp. Epidemiol. 2015, 36, 794–801.
- Huang, A.M.; Marini, B.L.; Frame, D.; Aronoff, D.M.; Nagel, J.L. Risk Factors for Recurrent Clostridium Difficile Infection in Hematopoietic Stem Cell Transplant Recipients. Transpl. Infect. Dis. 2014, 16, 744–750.
- Kelly, C.P.; Becker, S.; Linevsky, J.K.; Joshi, M.A.; O’Keane, J.C.; Dickey, B.F.; LaMont, J.T.; Pothoulakis, C. Neutrophil Recruitment in Clostridium Difficile Toxin A Enteritis in the Rabbit. J. Clin. Investig. 1994, 93, 1257–1265.
- Jose, S.; Mukherjee, A.; Abhyankar, M.M.; Leng, L.; Bucala, R.; Sharma, D.; Madan, R. Neutralization of Macrophage Migration Inhibitory Factor Improves Host Survival after Clostridium Difficile Infection. Anaerobe 2018, 53, 56–63.
- Kulaylat, A.S.; Buonomo, E.L.; Scully, K.W.; Hollenbeak, C.S.; Cook, H.; Petri, W.A.J.; Stewart, D.B.S. Development and Validation of a Prediction Model for Mortality and Adverse Outcomes Among Patients With Peripheral Eosinopenia on Admission for Clostridium Difficile Infection. JAMA Surg. 2018, 153, 1127–1133.
- Ryan, A.; Lynch, M.; Smith, S.M.; Amu, S.; Nel, H.J.; McCoy, C.E.; Dowling, J.K.; Draper, E.; O’Reilly, V.; McCarthy, C.; et al. A Role for TLR4 in Clostridium Difficile Infection and the Recognition of Surface Layer Proteins. PLoS Pathog. 2011, 7, e1002076.
- Jarchum, I.; Liu, M.; Lipuma, L.; Pamer, E.G. Toll-like Receptor 5 Stimulation Protects Mice from Acute Clostridium Difficile Colitis. Infect. Immun. 2011, 79, 1498–1503.
- Ng, J.; Hirota, S.A.; Gross, O.; Li, Y.; Ulke-Lemee, A.; Potentier, M.S.; Schenck, L.P.; Vilaysane, A.; Seamone, M.E.; Feng, H.; et al. Clostridium Difficile Toxin-Induced Inflammation and Intestinal Injury Are Mediated by the Inflammasome. Gastroenterology 2010, 139, 542–552, 552.e1-3.
- Liu, Y.-H.; Chang, Y.-C.; Chen, L.-K.; Su, P.-A.; Ko, W.-C.; Tsai, Y.-S.; Chen, Y.-H.; Lai, H.-C.; Wu, C.-Y.; Hung, Y.-P.; et al. The ATP-P2X(7) Signaling Axis Is an Essential Sentinel for Intracellular Clostridium Difficile Pathogen-Induced Inflammasome Activation. Front. Cell Infect. Microbiol. 2018, 8, 84.
- Collins, L.E.; Lynch, M.; Marszalowska, I.; Kristek, M.; Rochfort, K.; O’Connell, M.; Windle, H.; Kelleher, D.; Loscher, C.E. Surface Layer Proteins Isolated from Clostridium Difficile Induce Clearance Responses in Macrophages. Microbes Infect. 2014, 16, 391–400.
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066.
- Geiger, T.L.; Abt, M.C.; Gasteiger, G.; Firth, M.A.; O’Connor, M.H.; Geary, C.D.; O’Sullivan, T.E.; van den Brink, M.R.; Pamer, E.G.; Hanash, A.M.; et al. Nfil3 Is Crucial for Development of Innate Lymphoid Cells and Host Protection against Intestinal Pathogens. J. Exp. Med. 2014, 211, 1723–1731.
- Abt, M.C.; Lewis, B.B.; Caballero, S.; Xiong, H.; Carter, R.A.; Sušac, B.; Ling, L.; Leiner, I.; Pamer, E.G. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium Difficile Infection. Cell Host Microbe 2015, 18, 27–37.
- Hasegawa, M.; Yada, S.; Liu, M.Z.; Kamada, N.; Muñoz-Planillo, R.; Do, N.; Núñez, G.; Inohara, N. Interleukin-22 Regulates the Complement System to Promote Resistance against Pathobionts after Pathogen-Induced Intestinal Damage. Immunity 2014, 41, 620–632.
- Fachi, J.L.; Sécca, C.; Rodrigues, P.B.; Mato, F.C.P.D.; Di Luccia, B.; Felipe, J.D.S.; Pral, L.P.; Rungue, M.; Rocha, V.D.M.; Sato, F.T.; et al. Acetate Coordinates Neutrophil and ILC3 Responses against C. Difficile through FFAR2. J. Exp. Med. 2020, 217, e20190489.
- Chaplin, D.D. Overview of the Immune Response. J. Allergy Clin. Immunol. 2010, 125, S3–S23.
- Lessa, F.C.; Winston, L.G.; McDonald, L.C. Burden of Clostridium Difficile Infection in the United States. N. Engl. J. Med. 2015, 372, 2369–2370.
- Chilton, C.H.; Pickering, D.S.; Freeman, J. Microbiologic Factors Affecting Clostridium Difficile Recurrence. Clin. Microbiol. Infect. 2018, 24, 476–482.
- Leav, B.A.; Blair, B.; Leney, M.; Knauber, M.; Reilly, C.; Lowy, I.; Gerding, D.N.; Kelly, C.P.; Katchar, K.; Baxter, R.; et al. Serum Anti-Toxin B Antibody Correlates with Protection from Recurrent Clostridium Difficile Infection (CDI). Vaccine 2010, 28, 965–969.
- Johnson, S.; Gerding, D.N.; Janoff, E.N. Systemic and Mucosal Antibody Responses to Toxin A in Patients Infected with Clostridium Difficile. J. Infect. Dis. 1992, 166, 1287–1294.
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Asymptomatic Carriage of Clostridium Difficile and Serum Levels of IgG Antibody against Toxin A. N. Engl. J. Med. 2000, 342, 390–397.
- Warny, M.; Vaerman, J.P.; Avesani, V.; Delmée, M. Human Antibody Response to Clostridium Difficile Toxin A in Relation to Clinical Course of Infection. Infect. Immun. 1994, 62, 384–389.
- Bauer, M.P.; Nibbering, P.H.; Poxton, I.R.; Kuijper, E.J.; van Dissel, J.T. Humoral Immune Response as Predictor of Recurrence in Clostridium Difficile Infection. Clin. Microbiol. Infect. 2014, 20, 1323–1328.
- Law, H.; Venturi, V.; Kelleher, A.; Munier, C.M.L. Tfh Cells in Health and Immunity: Potential Targets for Systems Biology Approaches to Vaccination. Int. J. Mol. Sci. 2020, 21, 8524.
- Mintz, M.A.; Cyster, J.G. T Follicular Helper Cells in Germinal Center B Cell Selection and Lymphomagenesis. Immunol. Rev. 2020, 296, 48–61.
- Amadou Amani, S.; Shadid, T.; Ballard, J.D.; Lang, M.L. Clostridioides Difficile Infection Induces an Inferior IgG Response to That Induced by Immunization and Is Associated with a Lack of T Follicular Helper Cell and Memory B Cell Expansion. Infect. Immun. 2020, 88, 00829-19.
- Jafari, N.V.; Kuehne, S.A.; Bryant, C.E.; Elawad, M.; Wren, B.W.; Minton, N.P.; Allan, E.; Bajaj-Elliott, M. Clostridium Difficile Modulates Host Innate Immunity via Toxin-Independent and Dependent Mechanism(s). PLoS ONE 2013, 8, e69846.
- Hamo, Z.; Azrad, M.; Nitzan, O.; Peretz, A. Characterization of the Immune Response during Infection Caused by Clostridioides Difficile. Microorganisms 2019, 7, 435.
- Littmann, E.R.; Lee, J.-J.; Denny, J.E.; Alam, Z.; Maslanka, J.R.; Zarin, I.; Matsuda, R.; Carter, R.A.; Susac, B.; Saffern, M.S.; et al. Host Immunity Modulates the Efficacy of Microbiota Transplantation for Treatment of Clostridioides Difficile Infection. Nat. Commun. 2021, 12, 755.
This entry is offline, you can click here to edit this entry!