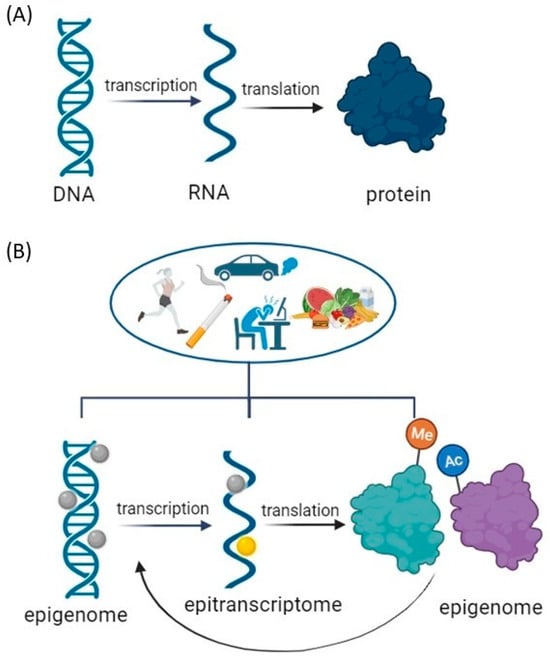
Epigenetic modifications to DNA and histones regulate gene transcription, whereas epitranscriptomic post-transcriptional RNA modifications influence gene expression [
165]. The most common mRNA modifications are N1-methyladenosine (m
1A), N6-methyladenosine (m
6A), 5-methylcytosine (m
5C), pseudouridine (Ψ), and others [
166]. m
6A modification is one of the most abundant and reversible epitranscriptomic modifications, mediated by a set of proteins, which include methyltransferases (‘writers’), demethylases (‘erasers’), and m
6A-binding proteins (‘readers’) [
167]. m
6A methylation is associated with the control of mRNA metabolism, splicing, export, stability, translation, and degradation [
168,
169]. Out of all tissues in the body, the brain has the highest abundance of m
6A methylation, which is developmentally decreasing [
168]. In the brain, m
6A methylation regulates neuronal transcripts and neuronal activity. Aside from its role in neuronal development [
30], m
6A modification is essential for the process of axon regeneration [
170]. Methyltransferases like-3 (METTL3) and like-14 (METTL14) [
171,
172,
173], along with other proteins needed for m
6A deposition, such as Wilms’ tumor 1-associating protein (WTAP) [
174] and RNA binding protein 15 (Rbm15) [
175], form a stable protein complex that catalyzes m
6A modification. Because they deposit RNA methylation modifications, these methyltransferases are collectively known as “writers”. Fat mass obesity-associated protein (FTO) and alkB homologue 5 (ALKBH5) are two demethylases from the family of α-ketoglutarate dependent dioxygenases that can reverse m
6A modification because it is dynamically regulated [
176,
177]. Demethylases are known as “erasers” because they remove RNA methylation modifications. Posttranscriptional, site-specific adenosine-to-inosine base conversions, known as RNA editing, contribute to gene expression diversity and are catalyzed by Adenosine deaminases acting on RNA (ADARs) [
178]. Pseudouridine Synthase 7 (PUS7) is one of the major mRNA-modifying enzymes leading to pseudouridine (Ψ), a ubiquitous RNA modification [
179]. Both ADAR and PUS7 can lead to further mRNA modifications. m
6A modifications are in direct connection to the so-called m
6A “reader” proteins, which recognize the modified site. The proteins with YTH domains, which can specifically bind m
6A through their YTH domain, are the most well-studied m
6A readers. Fragile X mental retardation protein (FMRP) was also reported to be m
6A reader and plays critical roles in synaptic plasticity and neuronal development. The identification of methylated nucleosides (m
6A, m
5C, m
1A) is performed using immunoprecipitation and their variants using antibodies against methylated nucleosides or associated proteins methyltransferases, demethylases, and binding proteins. After fragmenting the RNA, fragments containing modified nucleosides are enriched before sequencing. The immunoprecipitate is analyzed using next-generation sequencing (NGS) to identify and map the modification [
180].
The connection between epigenetic regulation and m
6A RNA modification was associated with histone H3 trimethylation at Lys36 (H3K36me3), a marker for transcription elongation, which guides m
6A deposition globally connected through METTL14 (DOI: 10.1038/s41586-019-1016-7). m
6A modifications are mainly associated with neuronal plasticity in the brain, which is a consequence of learning and memory, and most of the literature is based on this line of research together with neurodegenerative disorders [
176,
181,
182]. Indeed, deficiency in m
6A-dependent pathways significantly impairs neuronal function including dopamine signaling and dopamine-dependent learning.
Lowering neuronal m
6A by overexpressing FTO or by adding m
6A inhibitor led to the induction of N-methyl-d-aspartate (NMDA) receptor 1 expression, elevated oxidative stress, and Ca
2+ influx, resulting in dopaminergic neuron apoptosis [
183]. In addition, it was shown that the overexpression of FTO delays the dephosphorylation of CREB, increases the expression of the CREB, and targets neuropeptide receptor 1 (NPY1R) and BDNF known to regulate food intake and energy homeostasis [
184]. FTO affects dopamine (D2)-dependent responses to reward learning in meso-striato-prefrontal regions, suggesting a mechanism by which genetic predisposition alters reward processing not only in obesity but also in other disorders with altered D2R-dependent impulse control, such as addiction [
185].