Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Retinopathy of prematurity (ROP) is a proliferative vascular ailment affecting the retina. It is the main risk factor for visual impairment and blindness in infants and young children worldwide. If left undiagnosed and untreated, it can progress to retinal detachment and severe visual impairment. Geographical variations in ROP epidemiology have emerged over recent decades, attributable to differing levels of care provided to preterm infants across countries and regions.
- retinopathy of prematurity
- pathophysiology
- signaling pathways
- novel
- molecular targets
1. Introduction
Retinopathy of prematurity (ROP) is a developmental vascular proliferative disease affecting the retina characterized by abnormal capillary growth in infants born preterm [1]. Key risk factors encompass low gestational age, low birth weight, and the utilization of prolonged mechanical, and particularly fluctuating, ventilation, often employed as a therapeutic measure in preterm infants [2][3][4]. The surge in premature births globally has led to a dramatically increased incidence of ROP. If left undetected and untreated, it can result in severe consequences for the visual system, including retinal detachment and irreversible vision impairment. ROP ranks among the primary causes of childhood blindness worldwide [5].
In the 1940s and 1950s, the unrestrained use of oxygen (O2) in industrialized nations sparked the initial epidemic of retinal disease in premature births, termed retrolental fibroplasia [6][7][8][9]. Subsequently, advancements in neonatal care and perinatal monitoring in the 1960s and 1970s increased the survival rate of extremely premature infants, causing a second surge in retinal disease [10][11]. This trend continued with a third peak affecting middle-income countries and regions like China, Southeast Asia, South Asia, South America, and Eastern Europe due to enhanced survival rates of very premature infants through improved neonatal care [12][13][14][15][16][17][18][19][20]. More recently, European countries and the US have witnessed a rising trend in ROP incidence. For instance, a study in the UK revealed a 4% incidence of ROP requiring treatment among preterm babies weighing < 1500 g [21]. Similarly, studies in Greece and Norway reported incidences of around 18.4% and 39.6%, respectively, among preterm infants [22]. In the US, the incidence rose from 11% in 2009 to 15% in 2018 among neonates meeting ROP screening criteria [23]. Currently, developed regions like the US and the UK tend to show a lower incidence, while developing regions such as India and Africa demonstrate slightly higher ROP incidence rates [24].
Despite advancements in ROP research and treatment, its incidence continues to rise, leading to escalated healthcare costs. Gyllensten et al. conducted a meta-analysis highlighting that the costs of ROP screening (ranging between USD 324–USD 1072 per child) and therapy (ranging between USD 38–USD 6500 per child) are considerably lower than the societal costs of resulting blindness (ranging between USD 26,686–USD 224,295), emphasizing the pivotal role of screening tools in combating this disorder [25]. Similarly, a cost-effectiveness analysis in Mexico and the US estimated substantial annual benefits of around USD 206 million and USD 205 million, respectively, by implementing national ROP screening and treatment programs [26]. Another systematic review reaffirmed the high cost-effectiveness of ROP screening and treatment in the UK, Canada, and the US [27].
2. Retinal Development and Disease Pathogenesis
During physiological retinal angiogenesis, blood vessels begin to form around the 14–15th week of gestation, originating from the optic nerve head and expanding centrifugally towards the retinal periphery [28]. By 36 weeks of gestation, the nasal portion of the retina becomes vascularized, while the temporal area completes this process by the 40th week. Consequently, preterm infants exhibit incompletely vascularized retinas, with the extent of the avascular zone contingent upon their gestational age [29].
Under normal circumstances, the hypoxic conditions typical of the intrauterine environment stimulate retinal vascularization by prompting the expression of hypoxia-inducible factor 1α (HIF-1α). This factor regulates the expression of various oxygen-sensing genes, including crucial proangiogenic factors like vascular endothelial growth factor (VEGF) [30]. Although the VEGF family encompasses several members, including VEGF-A, -B, -C, -D, and placental growth factor (PlGF), it is VEGF-A that predominantly drives retinal angiogenesis [31][32].
VEGF, primarily released by neuroglia, initiates retinal blood vessel formation through the migration of vascular endothelial cells in a paracrine manner [33]. As retinal angiogenesis progresses, hypoxic conditions diminish, leading to the cessation of HIF-1 activation and its target genes. However, premature exposure to oxygen (hyperoxia) in an immature retina significantly suppresses HIF-1 and VEGF activity, resulting in oxidative stress and the emergence of avascular retinal regions [28]. Nitro-oxidative stress, characterized by an imbalance between the abundant generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and antioxidative defense mechanisms, leads to excess ROS and RNS. This imbalance triggers dramatic structural molecular changes and the activation of inflammatory and cell death pathways, a condition detectable in various eye diseases [34][35]. It is noteworthy that infants possess reduced antioxidant defenses [36]. In this context, Buhimish and colleagues reported that preterm births exhibit a lack of compensatory upregulation of nonenzymatic antioxidant reserves, such as glutathione (GSH) and plasma total free radical-trapping antioxidant potential [37]. Given the diminished antioxidative capability and increased vulnerability to oxidative stress in preterm neonates, it is not surprising that several oxidative biomarkers, such as malondialdehyde (MDA), 8-hydroxy 2-deoxyguanosine (8-OHdG), and the GSH/GSSG ratio, have been discussed as potential diagnostic tools for ROP [38].
An overabundance of ROS is described during the first phase of ROP, a status of hyperoxia that manifests as vaso-obliteration, characterized by decreased levels of HIF-1α, VEGF, and IGF-1 [39]. Subsequently, an ischemic phase ensues, gradually progressing into a proliferative stage marked by abnormal and dysfunctional neoangiogenesis. This phase ultimately results in intravitreal fibrosis, retinal traction, and detachment [28][39].
Ashton et al. introduced the two-phase hypothesis on ROP pathogenesis, demonstrating that exposing healthy cats to 70–80% oxygen for four days induces newly formed capillaries, leading to a process of “vaso-obliteration.” Upon returning to normal air exposure, a phase of “vasoproliferation” is observed [40]. In the initial phase (phase I), physiological retinal angiogenesis is delayed due to high oxygen exposure, resulting in vascular occlusion, reduced serum IGF-1, and delayed expression of VEGF receptors 2 [41]. Subsequently (phase II), retinal and vitreous neoangiogenesis occurs alongside increased levels of HIF-1α, VEGF, IGF-1, placental growth factor, erythropoietin (EPO), metalloproteinase (MMP)-2, MMP-9, and angiopoietin (Ang)-2 [42]. Another model, the oxygen-induced retinopathy (OIR) mouse model, mimics high oxygen levels similar to Ashton’s experiments. In this model, exposure to constant high oxygen (75% O2) causes newly formed capillaries to regress, leading to central areas of vaso-obliteration. Upon returning to room air, relative hypoxia triggers the release of angiogenic factors, promoting the vasoproliferation of blood vessels into the vitreous [43].
Figure 1 illustrates Phases 1 and 2 in the pathogenesis of ROP, focusing on the distinct expression of key mediators contributing to the onset of the disease.
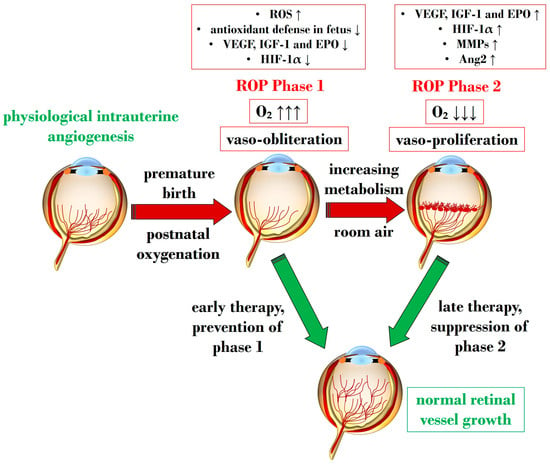
Figure 1. Schematic representation depicting the two pathogenetic phases in ROP, highlighting distinct expression levels of key mediators in the retina. ROP: retinopathy of prematurity; ROS: reactive oxygen species; O2: oxygen; VEGF: vascular endothelial growth factor; IGF-1: insulin-like growth factor 1; EPO: erythropoietin; HIF-1α: hypoxia-inducible factor 1 alpha; MMP: metalloproteinase; Ang-2: angiopoietin 2. Upward black arrows indicate upregulation or increased concentration, downward black arrows indicate downregulation or decreased concentration.
3. Exploring Molecular Cascades in Retinopathy of Prematurity
Throughout the various pathological stages of ROP, a multitude of molecular signaling pathways emerge, contributing to inflammatory processes and an abundance of ROS and RNS—two crucial factors in the early stages of ROP [29][44].
Subsequent sections delve into the primary pathways responsible for vaso-obliteration and vaso-proliferation during ROP pathogenesis.
3.1. The Central Role of Nitro-Oxidative Stress and Inflammatory Factors
In ROP, endothelial cell apoptosis triggered by oxidative stress is implicated in the process of vaso-obliteration that occurs in the retina during the initial phase of the disorder. Gu et al. demonstrated in bovine retinal endothelial cells that hyperoxia-induced nitro-oxidative stress leads to retinal capillary endothelial cell apoptosis, potentially by impeding growth factor-induced activation of the PI3K/Akt signaling pathway [45]. The pro-oxidative enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), present in isoforms NOX1, NOX2, and NOX4, generates high levels of superoxide (O2∙−), one of the most detrimental ROS, and has been associated with oxidative stress and vascular neoangiogenesis in OIR models [46][47][48]. Wang et al. reported in a rodent model of OIR that NOX4 might regulate intravitreal neovascularization mediated by VEGFR2 via activated signal transducer and activator of transcription (STAT) 3 within endothelial cells [47]. Studies by Saito et al. on animal models of ROP showcased that hyperoxia activates NOX, leading to an excess of ROS, culminating in apoptosis and neoangiogenesis independently of VEGF [49][50]. Furthermore, Byfield et al. demonstrated in a rodent model subjected to repeated oxygen fluctuations that hyperoxia-induced NOX activation leads to intravitreal VEGF-associated vascularization through a Janus tyrosine kinase (JAK)2/STAT3 pathway, and inhibiting this signaling pathway reduced neoangiogenesis [51]. However, VEGF signaling pathways seem to play a role in both phases of ROP, influencing pro-inflammatory and pro-oxidative processes. The same research group later revealed that VEGF-induced STAT3 activation blocked retinal angiogenesis by downregulating local expression of erythropoietin (EPO) in Müller cells during phase 1 of OIR [52]. Ren et al.’s investigation in a rodent model exposed to hyperoxia demonstrated that hyperoxia-induced STAT3 signaling enhances hepcidin expression—a key protein involved in iron balance—suggesting a potential compensatory mechanism to counteract iron overload linked to neoangiogenesis and proposing targeting molecules to regulate iron regulation [53].
Nitric oxide (NO) is pivotal in regulating vasodilation processes. Three main isoforms of NO synthases (NOS)—neural (n)NOS, inducible (i)NOS, and endothelial (e)NOS—are described in the literature. To counteract vaso-obliteration, vasodilation via NO synthesis occurs during the early phases of ROP [54]. However, differential NO responses are associated with the redox state [55]. As the disease progresses, excess NO synthesis becomes detrimental, favoring neoangiogenesis [56]. Importantly, normal NOS activity relies on the availability of the cofactor (6R)-5,6,7,8-tetrahydrobiopterin (BH4). Edgar et al. assessed in a murine model of OIR that exposure to oxygen led to decreased BH4 levels in the retinas, lungs, and aortas of mice, resulting in increased NOS-related ROS [57]. In circumstances of reduced BH4, uncoupled eNOS activity leads to nitro-oxidative stress in ROP pathogenesis. Specifically, in hyperoxia, impaired eNOS generates peroxynitrite (ONOO−) rather than physiological NO. Alongside eNOS, iNOS has also been reported as a pathogenetic factor in ROP. For instance, hypoxia-induced activation of iNOS in a murine model of OIR was linked to HIF-1α activation, VEGF expression, and PI3K/Akt signaling during neoangiogenesis, and inhibiting iNOS reduced the expression of these mediators [58]. However, NO also plays a significant role in neoangiogenesis processes during the vaso-proliferative phase, being fundamental in events of vascular permeability and leakage observed in retinopathies, affecting adherent junctions and endothelial cell polarity [59][60].
Notably, nitro-oxidative stress can interfere with prostanoid metabolism, exacerbating vaso-obliteration and contributing to avascular retinal onset. Reactive nitrogen species were observed in a murine OIR model to promote an isomerization of arachidonic acid to trans-arachidonic acid, involved in upregulating the anti-angiogenic factor thrombospondin-1 [61]. In this context, the role of phospholipase A2 (PLA-2) becomes noteworthy—an enzyme triggered by hypoxia and ROS abundance, influencing prostanoid metabolism via arachidonic acid release [62]. This molecule acts as a substrate for cyclooxygenase (COX), which converts it into proangiogenic eicosanoids, such as prostaglandins, prostacyclin, and thromboxane. Barnett et al. showed that suppressing PLA-2 decreased proangiogenic prostaglandins and intravitreal neoangiogenesis in an OIR model [62].
Arginase, present in isoforms arginase 1 and 2, hydrolyzes L-arginine to ornithine and urea, also producing glutamate [63]. This enzyme has been implicated in neural regeneration and protection [64]. Preterm infants exhibit low arginine levels, a crucial amino acid for retinal vascular development [65][66]. Arginine administration, particularly intravitreally with glutamine, countered neoangiogenesis in an OIR mouse model [67]. Importantly, arginase functionality and expression are heightened in contexts of inflammation and ROS excess, potentially interfering with NOS activity by competing for the substrate L-arginine. This competition indirectly triggers NOS uncoupling, leading to an overproduction of ONOO− [68][69]. Arginase 1 has been associated with neuroprotection [70], while arginase 2 might play a role in the events leading to retinal injuries, closely linked with nitro-oxidative stress and inflammation [71]. Shosha et al. proposed that NOX2-related O2∙− induces an upregulation of arginase 2 in ischemia/reperfusion injury, contributing to neurovascular degeneration [72].
In addition to oxidative stress, inflammatory events play a crucial role in the pathogenesis of ROP [73]. Cytokines such as IL-1β, tumor necrosis factor-α (TNF-α), and IL-6 are identified as primary drivers of inflammation, capable of inducing the overexpression of various inflammatory mediators, including chemokines and adhesion molecules. Specifically, within the hypoxic neonatal retina, retinal microglia can produce substantial amounts of IL-1β and TNF-α, ultimately promoting the death of retinal ganglion cells [74]. Furthermore, a study by Rivera and colleagues demonstrated in an OIR model that IL-1β is associated with retinal microvascular degeneration, triggering the release of the proapoptotic/repulsive factor semaphorin-3A from neurons [75]. Subsequent investigations by the same research group on the same model revealed the early pivotal role of IL-1β in the choroid, contributing to the involution of choroidal blood vessels and causing a loss of retinal pigment epithelium and photoreceptors [76]. As a consequence of cytokine downstream activation, chemokines also play a role in the pathogenesis of ROP, facilitating chemotaxis and the recruitment of immune cells to sites of inflammation. Specifically, chemokines implicated in ROP include IL-8, “RANTES” (Regulated and Normal T-cell Expressed and Secreted), and monocyte chemotactic protein 1 [77][78][79][80][81][82]. Taken together, inflammatory factors are pivotal in the pathophysiology of ROP, considering their role in orchestrating, together with oxidative stress, an amplification of the aberrant immune-mediated activation that leads to retinal cell death and choroidal degeneration.
3.2. The Crucial Involvement of HIF-1α and VEGF
HIF is a transcription factor composed of two subunits: HIF-1α (or its analogs HIF-2α and HIF-3α) and HIF-1β [83]. In normoxic conditions, HIF-1α undergoes hydroxylation by a prolyl hydroxylase domain (PHD) in the cytosol [84]. However, under hypoxic conditions, the enzymatic activities of PHD are inhibited, resulting in an increase in HIF-1α expression. Subsequently, HIF-1α binds to HIF-1β in the nucleus, forming the HIF-1 complex, which activates angiogenic mechanisms to help cells adapt to hypoxia. HIF-1α orchestrates the expression of several neoangiogenic mediators, including VEGF, EPO, angiopoietin (Ang)-1, and Ang-2, all observed to be upregulated in phase 2 of ROP [85]. Notably, under hypoxic conditions, HIF-1α is pivotal in reprogramming cellular metabolism, enhancing glycolysis, and increasing mitochondrial NADPH synthesis [86]. Even under normoxic conditions, HIF-1α activity can be induced by ROS and stabilized by inflammatory cytokines and growth factors like IGF-1 and TGF-β [87][88]. Enhanced HIF-1α activity due to relative intrauterine hypoxia is critical for physiological retinal vascular development [89]. However, in premature births, postnatal hyperoxia suppresses HIF-1α activity, leading to reduced VEGF release and consequent retinal capillary obliteration [90][91]. Stabilizing HIF-1α might represent a potential molecular target to halt the progression to phase 2 of ROP, as discussed in Section 4.2.4.
As mentioned earlier, VEGF plays a crucial role in both phases of ROP. Reduced VEGF levels under hyperoxia contribute to vaso-obliteration via endothelial cell apoptosis [92][93]. Conversely, in phase 2 under hypoxia, increased retinal VEGF levels act on endothelial cells through a paracrine route [94]. VEGF’s signaling involves the downregulation of retinal EPO in Müller cells via STAT3 activation. Furthermore, retinal endothelium expresses VEGFR-2, a receptor pivotal in neoangiogenic events and responsible for directing dividing endothelial cells in the developing retina [95]. Upregulation of VEGFR-2 disrupts dividing endothelial cells, potentially driving them to develop outside the retina, as observed in models of intravitreal neovascularization [96]. Inhibiting VEGFR-2 has shown promise in reducing intravitreal neoangiogenesis in preclinical investigations using the OIR model [97].
Figure 2 provides a schematic overview illustrating the primary molecular pathways that unfold during hyperoxia and hypoxia in ROP.
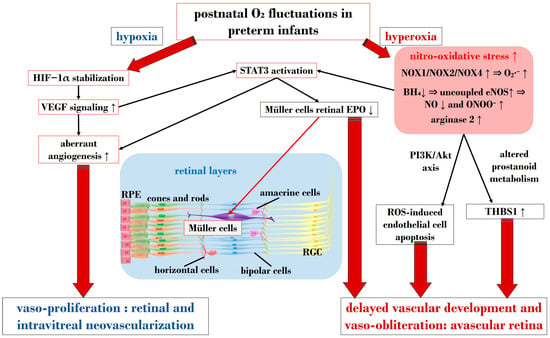
Figure 2. Schematic overview depicting the primary molecular pathways activated during phases 1 and 2 in ROP. ROS: reactive oxygen species; NOX: nicotinamide adenine dinucleotide phosphate oxidase; O2: oxygen; O2∙−: superoxide; ONOO−: peroxynitrite; VEGF: vascular endothelial growth factor; EPO: erythropoietin; HIF-1α: hypoxia-inducible factor 1 alpha; BH4: tetrahydrobiopterin; NO: nitric oxide; eNOS: endothelial nitric oxide synthase; STAT3: signal transducer and activator of transcription 3; RGC: retinal ganglion cell; RPE: retinal pigment epithelium; THBS1: thrombospondin 1. Upward black arrows indicate upregulation or increased concentration, downward black arrows indicate downregulation or decreased concentration.
This entry is adapted from the peer-reviewed paper 10.3390/antiox13020148
References
- Rowe, L.W.; Belamkar, A.; Antman, G.; Hajrasouliha, A.R.; Harris, A. Vascular imaging findings in retinopathy of prematurity. Acta Ophthalmol. 2023, 00, 1–21.
- Schaffer, D.B.; Palmer, E.A.; Plotsky, D.F.; Metz, H.S.; Flynn, J.T.; Tung, B.; Hardy, R.J.; Cryotherapy for Retinopathy of Prematurity Cooperative Group. Prognostic factors in the natural course of retinopathy of prematurity. Ophthalmology 1993, 100, 230–237.
- Bossi, E.; Koerner, F.; Flury, B.; Zulauf, M. Retinopathy of prematurity: A risk factor analysis with univariate and multivariate statistics. Helv. Paediatr. Acta 1984, 39, 307–317.
- Enomoto, H.; Miki, A.; Matsumiya, W.; Honda, S. Evaluation of oxygen supplementation status as a risk factor associated with the development of severe retinopathy of prematurity. Ophthalmologica 2015, 234, 135–138.
- Kim, S.J.; Port, A.D.; Swan, R.; Campbell, J.P.; Chan, R.V.P.; Chiang, M.F. Retinopathy of prematurity: A review of risk factors and their clinical significance. Surv. Ophthalmol. 2018, 63, 618–637.
- Patz, A. Retrolental fibroplasia. Surv. Ophthalmol. 1969, 14, 1–29.
- Patz, A. The role of oxygen in retrolental fibroplasia. Trans. Am. Ophthalmol. Soc. 1968, 66, 940–985.
- Patz, A.; Hoeck, L.E.; De La Cruz, E. Studies on the effect of high oxygen administration in retrolental fibroplasia. I. Nursery observations. Am. J. Ophthalmol. 1952, 35, 1248–1253.
- Campbell, K. Intensive oxygen therapy as a possible cause of retrolental fibroplasia; a clinical approach. Med. J. Aust. 1951, 2, 48–50.
- Gibson, D.L.; Sheps, S.B.; Uh, S.H.; Schechter, M.T.; McCormick, A.Q. Retinopathy of prematurity-induced blindness: Birth weight-specific survival and the new epidemic. Pediatrics 1990, 86, 405–412.
- Phelps, D.L. Retinopathy of prematurity: An estimate of vision loss in the United States—1979. Pediatrics 1981, 67, 924–925.
- Chen, Y.; Li, X. Characteristics of severe retinopathy of prematurity patients in China: A repeat of the first epidemic? Br. J. Ophthalmol. 2006, 90, 268–271.
- Phan, M.H.; Nguyen, P.N.; Reynolds, J.D. Incidence and severity of retinopathy of prematurity in Vietnam, a developing middle-income country. J. Pediatr. Ophthalmol. Strabismus 2003, 40, 208–212.
- Azad, R.V.; Chandra, P. Retinopathy of prematurity—Screening and management. J. Indian Med. Assoc. 2003, 101, 593–596.
- Jalali, S.; Anand, R.; Kumar, H.; Dogra, M.R.; Azad, R.; Gopal, L. Programme planning and screening strategy in retinopathy of prematurity. Indian J. Ophthalmol. 2003, 51, 89–99.
- Gilbert, C.; Fielder, A.; Gordillo, L.; Quinn, G.; Semiglia, R.; Visintin, P.; Zin, A. Characteristics of infants with severe retinopathy of prematurity in countries with low, moderate, and high levels of development: Implications for screening programs. Pediatrics 2005, 115, e518–e525.
- Lermann, V.L.; Fortes Filho, J.B.; Procianoy, R.S. The prevalence of retinopathy of prematurity in very low birth weight newborn infants. J. Pediatr. (Rio J) 2006, 82, 27–32.
- Gilbert, C.E.; Anderton, L.; Dandona, L.; Foster, A. Prevalence of visual impairment in children: A review of available data. Ophthalmic Epidemiol. 1999, 6, 73–82.
- Gilbert, C.; Foster, A. Childhood blindness in the context of VISION 2020—The right to sight. Bull. World Health Organ. 2001, 79, 227–232.
- Gilbert, C.; Rahi, J.; Eckstein, M.; O’Sullivan, J.; Foster, A. Retinopathy of prematurity in middle-income countries. Lancet 1997, 350, 12–14.
- Adams, G.G.; Bunce, C.; Xing, W.; Butler, L.; Long, V.; Reddy, A.; Dahlmann-Noor, A.H. Treatment trends for retinopathy of prematurity in the UK: Active surveillance study of infants at risk. BMJ Open 2017, 7, e013366.
- Moutzouri, S.; Haidich, A.B.; Seliniotaki, A.K.; Tsakalidis, C.; Soubasi, V.; Ziakas, N.; Mataftsi, A. Optimization of retinopathy of prematurity screening in a tertiary neonatal unit in Northern Greece based on 16-year data. J. Perinatol. 2022, 42, 365–370.
- Thangamathesvaran, L.; Wang, J.; Repka, M.X.; Scott, A.W. Trends in Retinopathy of Prematurity Care in the United States 2009–2018: A Nationwide Analysis Using National Inpatient Sample. Ophthalmol. Retin. 2023, 7, 360–366.
- Chow, S.C.; Lam, P.Y.; Lam, W.C.; Fung, N.S.K. The role of anti-vascular endothelial growth factor in treatment of retinopathy of prematurity-a current review. Eye 2022, 36, 1532–1545.
- Gyllensten, H.; Humayun, J.; Sjöbom, U.; Hellström, A.; Löfqvist, C. Costs associated with retinopathy of prematurity: A systematic review and meta-analysis. BMJ Open 2022, 12, e057864.
- Rothschild, M.I.; Russ, R.; Brennan, K.A.; Williams, C.J.; Berrones, D.; Patel, B.; Martinez-Castellanos, M.A.; Fernandes, A.; Hubbard, G.B.; Chan, R.V.P.; et al. The Economic Model of Retinopathy of Prematurity (EcROP) Screening and Treatment: Mexico and the United States. Am. J. Ophthalmol. 2016, 168, 110–121.
- Smith, A.F.; Sadeq, A.; Kinzel, E.; Bhambhwani, V. A Systematic Review of Economic Evaluations Conducted for Interventions to Screen, Treat, and Manage Retinopathy of Prematurity (ROP) in the United States, United Kingdom, and Canada. Ophthalmic Epidemiol. 2023, 30, 113–120.
- Lucchesi, M.; Marracci, S.; Amato, R.; Filippi, L.; Cammalleri, M.; Dal Monte, M. Neurosensory Alterations in Retinopathy of Prematurity: A Window to Neurological Impairments Associated to Preterm Birth. Biomedicines 2022, 10, 1603.
- Fevereiro-Martins, M.; Marques-Neves, C.; Guimarães, H.; Bicho, M. Retinopathy of prematurity: A review of pathophysiology and signaling pathways. Surv. Ophthalmol. 2023, 68, 175–210.
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133.
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264.
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611.
- Stone, J.; Itin, A.; Alon, T.; Pe’Er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747.
- Böhm, E.W.; Buonfiglio, F.; Voigt, A.M.; Bachmann, P.; Safi, T.; Pfeiffer, N.; Gericke, A. Oxidative stress in the eye and its role in the pathophysiology of ocular diseases. Redox Biol. 2023, 68, 102967.
- Buonfiglio, F.; Böhm, E.W.; Pfeiffer, N.; Gericke, A. Oxidative Stress: A Suitable Therapeutic Target for Optic Nerve Diseases? Antioxidants 2023, 12, 1465.
- Hartnett, M.E. Advances in understanding and management of retinopathy of prematurity. Surv. Ophthalmol. 2017, 62, 257–276.
- Buhimschi, I.A.; Buhimschi, C.S.; Pupkin, M.; Weiner, C.P. Beneficial impact of term labor: Nonenzymatic antioxidant reserve in the human fetus. Am. J. Obstet. Gynecol. 2003, 189, 181–188.
- Graziosi, A.; Perrotta, M.; Russo, D.; Gasparroni, G.; D’Egidio, C.; Marinelli, B.; Di Marzio, G.; Falconio, G.; Mastropasqua, L.; Li Volti, G.; et al. Oxidative Stress Markers and the Retinopathy of Prematurity. J. Clin. Med. 2020, 9, 2711.
- Hellström, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457.
- Ashton, N.; Ward, B.; Serpell, G. Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br. J. Ophthalmol. 1954, 38, 397–432.
- Simmons, A.B.; Bretz, C.A.; Wang, H.; Kunz, E.; Hajj, K.; Kennedy, C.; Yang, Z.; Suwanmanee, T.; Kafri, T.; Hartnett, M.E. Gene therapy knockdown of VEGFR2 in retinal endothelial cells to treat retinopathy. Angiogenesis 2018, 21, 751–764.
- Ryu, J. New aspects on the treatment of retinopathy of prematurity: Currently available therapies and emerging novel therapeutics. Int. J. Mol. Sci. 2022, 23, 8529.
- Smith, L.E.H.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.J.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111.
- Wang, H.; Zhang, S.X.; Hartnett, M.E. Signaling pathways triggered by oxidative stress that mediate features of severe retinopathy of prematurity. JAMA Ophthalmol. 2013, 131, 80–85.
- Gu, X.; El-Remessy, A.B.; Brooks, S.E.; Al-Shabrawey, M.; Tsai, N.-T.; Caldwell, R.B. Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am. J. Physiol.-Cell Physiol. 2003, 285, C546–C554.
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.M.; Cooper, M.E.; et al. NADPH Oxidase, NOX1, Mediates Vascular Injury in Ischemic Retinopathy. Antioxid. Redox Signal. 2013, 20, 2726–2740.
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241.
- Chan, E.C.; van Wijngaarden, P.; Liu, G.S.; Jiang, F.; Peshavariya, H.; Dusting, G.J. Involvement of Nox2 NADPH oxidase in retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7061–7067.
- Saito, Y.; Uppal, A.; Byfield, G.; Budd, S.; Hartnett, M.E. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1591–1598.
- Saito, Y.; Geisen, P.; Uppal, A.; Hartnett, M.E. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol. Vis. 2007, 13, 840–853.
- Byfield, G.; Budd, S.; Hartnett, M.E. The role of supplemental oxygen and JAK/STAT signaling in intravitreous neovascularization in a ROP rat model. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3360–3365.
- Wang, H.; Byfield, G.; Jiang, Y.; Smith, G.W.; McCloskey, M.; Hartnett, M.E. VEGF-Mediated STAT3 Activation Inhibits Retinal Vascularization by Down-Regulating Local Erythropoietin Expression. Am. J. Pathol. 2012, 180, 1243–1253.
- Ren, J.; Jiang, J.; Ou, W.; Luo, X.; Xiang, J.; Liu, G.; Huang, S.; He, L.; Gan, J.; Li, H.; et al. The Effect of STAT3 Signal Pathway Activation on Retinopathy of Prematurity. Front. Pediatr. 2021, 9, 638432.
- Cavallaro, G.; Filippi, L.; Bagnoli, P.; La Marca, G.; Cristofori, G.; Raffaeli, G.; Padrini, L.; Araimo, G.; Fumagalli, M.; Groppo, M.; et al. The pathophysiology of retinopathy of prematurity: An update of previous and recent knowledge. Acta Ophthalmol. 2014, 92, 2–20.
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 2007, 26, 205–238.
- Opatrilova, R.; Kubatka, P.; Caprnda, M.; Büsselberg, D.; Krasnik, V.; Vesely, P.; Saxena, S.; Ruia, S.; Mozos, I.; Rodrigo, L.; et al. Nitric oxide in the pathophysiology of retinopathy: Evidences from preclinical and clinical researches. Acta Ophthalmol. 2018, 96, 222–231.
- Edgar, K.S.; Matesanz, N.; Gardiner, T.A.; Katusic, Z.S.; McDonald, D.M. Hyperoxia depletes (6R)-5,6,7,8-tetrahydrobiopterin levels in the neonatal retina: Implications for nitric oxide synthase function in retinopathy. Am. J. Pathol. 2015, 185, 1769–1782.
- He, T.; Ai, M.; Zhao, X.H.; Xing, Y.Q. Inducible nitric oxide synthase mediates hypoxia-induced hypoxia-inducible factor-1 alpha activation and vascular endothelial growth factor expression in oxygen-induced retinopathy. Pathobiology 2007, 74, 336–343.
- Smith, T.L.; Oubaha, M.; Cagnone, G.; Boscher, C.; Kim, J.S.; El Bakkouri, Y.; Zhang, Y.; Chidiac, R.; Corriveau, J.; Delisle, C.; et al. eNOS controls angiogenic sprouting and retinal neovascularization through the regulation of endothelial cell polarity. Cell. Mol. Life Sci. 2021, 79, 37.
- Ninchoji, T.; Love, D.T.; Smith, R.O.; Hedlund, M.; Vestweber, D.; Sessa, W.C.; Claesson-Welsh, L. eNOS-induced vascular barrier disruption in retinopathy by c-Src activation and tyrosine phosphorylation of VE-cadherin. Elife 2021, 10, e64944.
- Kermorvant-Duchemin, E.; Sennlaub, F.; Sirinyan, M.; Brault, S.; Andelfinger, G.; Kooli, A.; Germain, S.; Ong, H.; d’Orleans-Juste, P.; Gobeil, F., Jr. Trans-arachidonic acids generated during nitrative stress induce a thrombospondin-1–dependent microvascular degeneration. Nat. Med. 2005, 11, 1339–1345.
- Barnett, J.M.; McCollum, G.W.; Penn, J.S. Role of cytosolic phospholipase A(2) in retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1136–1142.
- Narayanan, S.P.; Rojas, M.; Suwanpradid, J.; Toque, H.A.; Caldwell, R.W.; Caldwell, R.B. Arginase in retinopathy. Prog. Retin. Eye Res. 2013, 36, 260–280.
- Lange, P.S.; Langley, B.; Lu, P.; Ratan, R.R. Novel roles for arginase in cell survival, regeneration, and translation in the central nervous system. J. Nutr. 2004, 134, 2812S–2817S.
- Fouda, A.Y.; Eldahshan, W.; Narayanan, S.P.; Caldwell, R.W.; Caldwell, R.B. Arginase pathway in acute retina and brain injury: Therapeutic opportunities and unexplored avenues. Front. Pharmacol. 2020, 11, 277.
- Tomita, Y.; Usui-Ouchi, A.; Nilsson, A.K.; Yang, J.; Ko, M.; Hellström, A.; Fu, Z. Metabolism in retinopathy of prematurity. Life 2021, 11, 1119.
- Neu, J.; Afzal, A.; Pan, H.; Gallego, E.; Li, N.; Calzi, S.L.; Caballero, S.; Spoerri, P.E.; Shaw, L.C.; Grant, M.B. The dipeptide Arg-Gln inhibits retinal neovascularization in the mouse model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3151–3155.
- Bachetti, T.; Comini, L.; Francolini, G.; Bastianon, D.; Valetti, B.; Cadei, M.; Grigolato, P.; Suzuki, H.; Finazzi, D.; Albertini, A. Arginase pathway in human endothelial cells in pathophysiological conditions. J. Mol. Cell. Cardiol. 2004, 37, 515–523.
- Suwanpradid, J.; Rojas, M.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency prevents oxidative stress and limits hyperoxia-induced retinal vascular degeneration. PLoS ONE 2014, 9, e110604.
- Fouda, A.Y.; Xu, Z.; Shosha, E.; Lemtalsi, T.; Chen, J.; Toque, H.A.; Tritz, R.; Cui, X.; Stansfield, B.K.; Huo, Y. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell Death Dis. 2018, 9, 1001.
- Shosha, E.; Fouda, A.Y.; Narayanan, S.P.; Caldwell, R.W.; Caldwell, R.B. Is the arginase pathway a novel therapeutic avenue for diabetic retinopathy? J. Clin. Med. 2020, 9, 425.
- Shosha, E.; Xu, Z.; Yokota, H.; Saul, A.; Rojas, M.; Caldwell, R.W.; Caldwell, R.B.; Narayanan, S.P. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016, 7, e2483.
- Rivera, J.C.; Holm, M.; Austeng, D.; Morken, T.S.; Zhou, T.E.; Beaudry-Richard, A.; Sierra, E.M.; Dammann, O.; Chemtob, S. Retinopathy of prematurity: Inflammation, choroidal degeneration, and novel promising therapeutic strategies. J. Neuroinflamm. 2017, 14, 165.
- Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.A.; Kaur, C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J. Pathol. 2011, 224, 245–260.
- Rivera, J.C.; Sitaras, N.; Noueihed, B.; Hamel, D.; Madaan, A.; Zhou, T.; Honoré, J.C.; Quiniou, C.; Joyal, J.S.; Hardy, P.; et al. Microglia and interleukin-1β in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1881–1891.
- Zhou, T.E.; Rivera, J.C.; Bhosle, V.K.; Lahaie, I.; Shao, Z.; Tahiri, H.; Zhu, T.; Polosa, A.; Dorfman, A.; Beaudry-Richard, A.; et al. Choroidal Involution Is Associated with a Progressive Degeneration of the Outer Retinal Function in a Model of Retinopathy of Prematurity: Early Role for IL-1β. Am. J. Pathol. 2016, 186, 3100–3116.
- Sullivan, G.; Galdi, P.; Cabez, M.B.; Borbye-Lorenzen, N.; Stoye, D.Q.; Lamb, G.J.; Evans, M.J.; Quigley, A.J.; Thrippleton, M.J.; Skogstrand, K.; et al. Interleukin-8 dysregulation is implicated in brain dysmaturation following preterm birth. Brain Behav. Immun. 2020, 90, 311–318.
- Silveira, R.C.; Fortes Filho, J.B.; Procianoy, R.S. Assessment of the contribution of cytokine plasma levels to detect retinopathy of prematurity in very low birth weight infants. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1297–1301.
- Powers, M.R.; Davies, M.H.; Eubanks, J.P. Increased expression of chemokine KC, an interleukin-8 homologue, in a model of oxygen-induced retinopathy. Curr. Eye Res. 2005, 30, 299–307.
- Hellgren, G.; Willett, K.; Engstrom, E.; Thorsen, P.; Hougaard, D.M.; Jacobsson, B.; Hellstrom, A.; Lofqvist, C. Proliferative retinopathy is associated with impaired increase in BDNF and RANTES expression levels after preterm birth. Neonatology 2010, 98, 409–418.
- Yoshida, S.; Yoshida, A.; Ishibashi, T.; Elner, S.G.; Elner, V.M. Role of MCP-1 and MIP-1α in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J. Leucoc. Biol. 2003, 73, 137–144.
- Yoshida, S.; Yoshida, A.; Ishibashi, T. Induction of IL-8, MCP-1, and bFGF by TNF-α in retinal glial cells: Implications for retinal neovascularization during post-ischemic inflammation. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 409–413.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Pazos-Sanou, L.; Mata-Segreda, J.F. Effect of ionic charge on detergent-induced hemolysis. Acta Physiol. Pharmacol. Latinoam. 1989, 39, 27–31.
- Hashimoto, T.; Shibasaki, F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015, 3, 33.
- Singh, C.; Hoppe, G.; Tran, V.; McCollum, L.; Bolok, Y.; Song, W.; Sharma, A.; Brunengraber, H.; Sears, J.E. Serine and 1-carbon metabolism are required for HIF-mediated protection against retinopathy of prematurity. JCI Insight 2019, 4, e129398.
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1) α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12.
- Cash, T.P.; Pan, Y.; Simon, M.C. Reactive oxygen species and cellular oxygen sensing. Free. Radic. Biol. Med. 2007, 43, 1219–1225.
- Weidemann, A.; Krohne, T.U.; Aguilar, E.; Kurihara, T.; Takeda, N.; Dorrell, M.I.; Simon, M.C.; Haase, V.H.; Friedlander, M.; Johnson, R.S. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 2010, 58, 1177–1185.
- Arjamaa, O.; Nikinmaa, M. Oxygen-dependent diseases in the retina: Role of hypoxia-inducible factors. Exp. Eye Res. 2006, 83, 473–483.
- Smith, L.E. Pathogenesis of retinopathy of prematurity. Growth Horm. IGF Res. 2004, 14, 140–144.
- Shih, S.-C.; Ju, M.; Liu, N.; Smith, L.E. Selective stimulation of VEGFR-1 prevents oxygen-induced retinal vascular degeneration in retinopathy of prematurity. J. Clin. Investig. 2003, 112, 50–57.
- Pierce, E.A.; Foley, E.D.; Smith, L.E. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch. Ophthalmol. 1996, 114, 1219–1228.
- Pierce, E.A.; Avery, R.L.; Foley, E.D.; Aiello, L.P.; Smith, L. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc. Natl. Acad. Sci. USA 1995, 92, 905–909.
- Ramshekar, A.; Hartnett, M.E. Vascular Endothelial Growth Factor Signaling in Models of Oxygen-Induced Retinopathy: Insights into Mechanisms of Pathology in Retinopathy of Prematurity. Front. Pediatr. 2021, 9, 796143.
- Hartnett, M.E.; Martiniuk, D.; Byfield, G.; Geisen, P.; Zeng, G.; Bautch, V.L. Neutralizing VEGF decreases tortuosity and alters endothelial cell division orientation in arterioles and veins in a rat model of ROP: Relevance to plus disease. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3107–3114.
- Budd, S.; Byfield, G.; Martiniuk, D.; Geisen, P.; Hartnett, M.E. Reduction in endothelial tip cell filopodia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp. Eye Res. 2009, 89, 718–727.
This entry is offline, you can click here to edit this entry!