Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Leukodystrophies, a group of rare demyelinating disorders, mainly affect the CNS. Clinical presentation of different types of leukodystrophies can be nonspecific, and thus, imaging techniques like MRI can be used for a more definitive diagnosis.
- leukodystrophies
- demyelinating disorders
- magnetic resonance imaging
- myelin imaging
1. Introduction
Leukodystrophies, a group of rare and debilitating disorders affecting the white matter of the central nervous system, present a complex interplay between pathology and genetics.
From advancements in genetic testing methodologies to the identification of novel genes associated with specific leukodystrophy subtypes, the genetic landscape of these disorders is rapidly expanding. This synthesis of the latest research in pathology, genetics, and imaging aims to offer a robust resource for clinicians, researchers, and genetic counselors striving for a deeper understanding of leukodystrophies and the development of targeted therapeutic interventions.
Magnetic resonance imaging (MRI) is the primary imaging technique to identify, localize, and characterize cerebral lesions in patients with leukodystrophy. These disorders pose a threat to the integrity of the brain and peripheral nerves, with clinical presentations often being nonspecific [1,2]. Imaging techniques, particularly MRI, play a crucial role in establishing a definitive diagnosis [3]. Current research suggests that early detection of leukodystrophy allows for more optimal implementation of therapy treatments, highlighting the importance of early disease detection [3,4].
Quantitative MRI, providing insights into myelin and axonal content, condition, and white matter composition, aids not only in diagnosis, but also in understanding disease progression [5]. Additionally, genetic testing, focusing on distinct alterations in specific genes, complements the diagnostic process, with advanced sequencing significantly accelerating the speed of diagnosis [6,7,8].
2. Types of Leukodystrophies
2.1. X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD), an X-linked genetic condition impacting the central and peripheral nervous systems along with the adrenal cortex, primarily manifests in boys and clinically presents with adrenal insufficiency, dysarthria, dysgraphia, vision, and hearing deficits, as well as neurocognitive and neurobehavioral issues [9,10]. The condition is marked by a mutation in the ABCD1 gene on the X chromosome, which is responsible for encoding the ALD protein, a transmembrane protein crucial for the transport of very long chain fatty acid (VLCFA)-CoA esters into the peroxisome [11,12]. Consequently, mutations in the ABCD1 gene lead to diminished VLCFA transport and widespread accumulation throughout the body [13]. Adrenocortical insufficiency typically develops in nearly all affected males around age 7.5, with the onset of progressive myelopathy and peripheral neuropathy occurring in adulthood [9,11]. Sixty percent of male patients will experience progressive and often fatal cerebral demyelination, a phenomenon detectable via the instrumental use of MRI, facilitating early detection and improving patient outcomes [14,15].
Newborn screening for X-ALD has gained widespread recommendation for implementation into the uniform screening panel in the United States, with over half of the states initiating screening. The diagnosis of X-ALD requires the detection of the ABCD1 pathogenic variant and the accumulation of VLCFAs [9]. Cerebral X-ALD cases present with distinctive white matter demyelination and inflammation, visualized on MRI, with lesions categorized into three zones [11,16,17]. These zones delineate the loss of axons, oligodendrocytes, and myelin sheaths, highlighting gadolinium enhancement and active macrophage involvement. Brain MRIs exhibit hyperintense confluent lesions of the corpus callosum and parieto-occipital white matter, progressing to impact the entire cerebral white matter in adulthood [18,19,20,21,22]. Routine brain MRIs are conducted to monitor disease progression, contrasting with the less utilized spinal cord imaging, which demonstrates corticospinal tract and dorsal column degeneration [23,24,25]. Furthermore, spinal cord MRI demonstrates corticospinal tract and dorsal column degeneration, resulting in the appearance of a flattened spinal cord and the reduction in anteroposterior diameters [23,24,25]. The pathophysiology of X-ALD, succinctly captured in Figure 1, underlines the prevention of VLCFA entry into the peroxisome due to ABCD1 gene mutations, resulting in the clinical phenotype arising from VLCFA accumulation throughout the body.
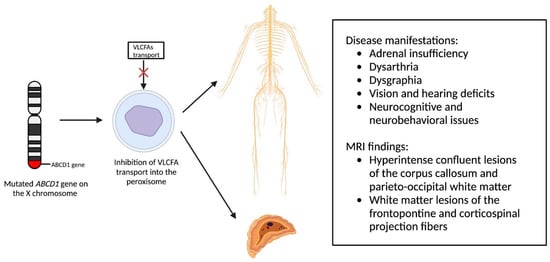
Figure 1. Pathophysiology of X-Linked Adrenoleukodystrophy: Mutations in the ABCD1 gene on the X chromosome result in the prevention of very long chain fatty acids (VLCFAs) into the peroxisome, resulting in the clinical phenotype due to VLCFA accumulation throughout the body.
While MRI remains the gold standard for lesion detection in X-ALD, emerging studies in other neurodegenerative disorders suggest the potential utility of Neurite Orientation Dispersion and Density Imaging (NODDI) in revealing increased orientation dispersion and a higher surface area of neurodegeneration compared to structural MRI and diffusion tensor imaging [26,27]. Additionally, myelin water fraction (MWF) imaging, assessing the quantity of myelin via specific water pools, presents a promising avenue for detecting early X-ALD lesions in the future [28].
2.2. Metachromatic Leukodystrophy
Metachromatic leukodystrophy, an autosomal recessive lysosomal storage disease, is characterized by a deficiency of arylsulfatase A (ARSA) due to a mutation in the arylsulfatase A gene on chromosome 22q13.3-qter [29,30]. ARSA plays a crucial role in the degradation of sulphatide, a membrane lipid found in myelin, the distal tubules of the kidney, and bile duct epithelia [29]. The deficiency of ARSA leads to the accumulation of sulphatide primarily affecting the nervous system and resulting in progressive demyelination, presenting clinically with ataxia, optic atrophy, dementia, and decerebrate posturing [29,31].
Children suspected of metachromatic leukodystrophy often exhibit delays in meeting developmental milestones and a decline in both gross and fine motor skills [30]. Diagnosis involves laboratory studies to assess ARSA levels, with criteria ranging from undetectable to less than 10% of the normal value [10]. Distinguishing metachromatic leukodystrophy from arylsulfatase A pseudodeficiency, present in about 1% of the general population, requires additional assessments such as urine sulfatide levels, radiolabeled sulfatide fibroblast loading, and DNA analysis.
As a demyelinating condition, MRI reveals brain demyelination and abnormalities in nerve conduction [32,33]. Initial impact occurs in the central and periventricular white matter, progressing to subcortical structures. Extreme cases may exhibit projection fiber involvement, leading to distinctive patterns like the “tigroid pattern” and “leopard skin” pattern as shown in Figure 2 [34,35]. Nearly all patients with metachromatic leukodystrophy show splenial corpus callosum demyelination [36]. T2-weighted FLAIR images display symmetric, confluent hyperintense areas in the periventricular white matter, consistent with the demyelinating nature of the disorder [37]. Lastly, MRI modalities, including diffusion-weighted parameters, demyelination load, and MR spectroscopy, hold potential in aiding early diagnosis by providing insights into nonspecific white matter changes associated with metachromatic leukodystrophy [38,39,40].
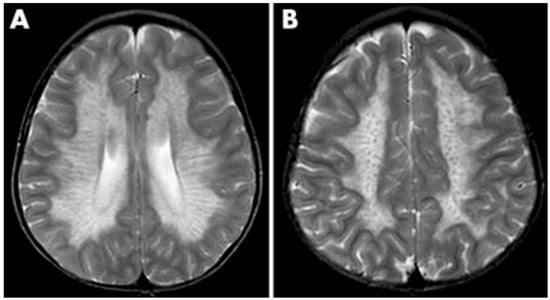
Figure 2. MRI findings found in metachromatic leukodystrophy. (A) Hypointense radial stripes resembling tiger skin. (B) Hypointense dots resembling leopard skin.
2.3. Krabbe’s Disease
Krabbe disease (KD), an inherited lysosomal storage disease, poses a diagnostic challenge due to its diverse clinical presentation and overlap with other neurodegenerative disorders [41]. This disease, also known as Globoid Cell Leukodystrophy, results from a defect in the GALC gene, leading to the accumulation of toxic myelin products [42]. KD typically manifests in infants under the age of six but can also occur in adolescents or adults, presenting symptoms such as muscle weakness, spasticity, hypertonia, myoclonic seizures, and sensory deficits [42,43]. At the presymptomatic stage, stem cell transplant can improve patient prognosis; however, without a family history of KD, the disorder is often not identified until the symptomatic stage. Once symptomatic, the patient becomes ineligible for transplantation [44]. For this reason, the goal of care is to screen infants for KD and identify it before symptoms appear.
Infant screening for KD is limited across the U.S., highlighting the importance of early identification before symptom onset [45]. Consensus guidelines recommend a three-step screening, diagnosis, and treatment process, which is detailed in Figure 3 [44]. The initial step involves a dried blood spot assay for GALC enzyme activity, followed by diagnostic tests at a specialty care center (SCC). Neurodiagnostic studies, including MRI and CSF analysis, are conducted at a human stem cell transplantation center (HSCT) upon confirmation of the diagnosis.
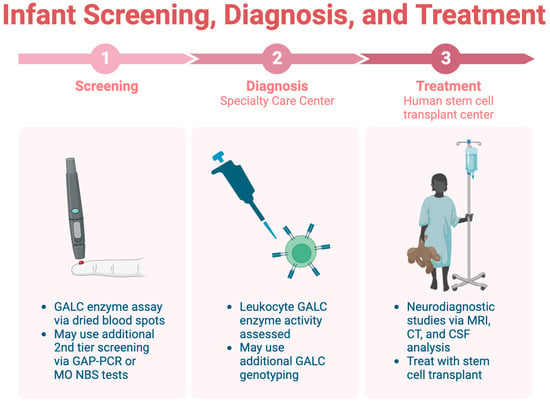
Figure 3. The three-step process for infant screening for KD.
The MRI findings in KD exhibit distinctive patterns across different subtypes, such as optic nerve and cervical cord enlargement in the infantile form and T2-hyperintense changes in corticospinal tracts in the adult form [46,47,48]. Psychosine accumulation in the CSF serves as a diagnostic tool, offering potential insights into gene therapy targets [47,49,50]. Despite its severity, KD diagnosis relies on a combination of clinical, genetic, and imaging assessments, emphasizing the significance of early screening and multidisciplinary approaches for optimal patient management.
2.4. Pelizaeus–Merzbacher Disease
Pelizaeus–Merzbacher disease (PMD) stands out as a rare leukodystrophy and CNS demyelinating disease, with its clinical manifestation documented by Friederich Pelizaeus and Ludwig Merzbacher in 1885 [51]. This neurological disorder presents symptoms such as nystagmus, spastic paralysis, and ataxia, leading to a progressive decline in coordination, motor skills, and cognitive function [52]. The X-linked recessive inheritance of PMD is attributed to mutations in the proteolipid protein 1 (PLP1) gene on the X chromosome, resulting in varied levels of decreased myelin production [51]. Classification into Types I, II, and III is contingent on the specific mutation, with Type I being the most severe [51].
Diagnosing PMD poses complexity due to overlapping symptoms with other leukodystrophies, necessitating the exclusion of alternative diagnoses [51]. Neonatal onset presents a more severe prognosis compared to the nearly benign adult form [53]. Inoue et al. introduced an interphase fluorescence in situ hybridization (FISH) assay for efficient screening, successfully diagnosing PMD and detecting carriers [54]. Molecular analysis further aids in identifying the size and location of gene duplications. Imaging via MRIs reveals gliosis around demyelinated areas, with T1-weighted sections indicating hypointensity and T2-weighted images displaying hyperintensity, reflecting demyelination (Figure 4). Magnetic resonance spectroscopy (MRS) findings in PMD cases vary, necessitating additional research for conclusive diagnostic utility [55,56].
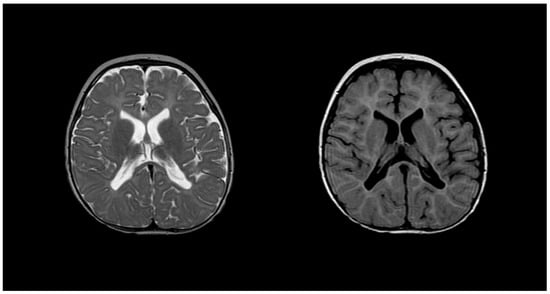
Figure 4. T2 (left) and T1 (right) imaging of a patient with Pelizaeus–Merzbacher disease showing lack of myelination in internal capsule, proximal corona radiata, and the optic radiation.
While FISH, MRI, and CSF analysis contribute to PMD identification, the reliance on exclusionary diagnosis due to overlapping findings underscores the need for further research. Current treatments focus on supportive care to alleviate symptoms, emphasizing the imperative for ongoing research to enhance the diagnosis and treatment of PMD and ultimately improve patients’ quality of life.
This entry is adapted from the peer-reviewed paper 10.3390/medsci12010007
This entry is offline, you can click here to edit this entry!