Ever since dopamine (DA) and norepinephrine (NE) neuronal pathways were identified and functionally characterized in vivo [
1,
2,
3,
4,
5,
6,
7,
8], the depth and breadth of studies of how these neurotransmitters affect both cognitive and motor behavior has been immense. The viability and function of the neuronal pathways that produce these neurotransmitters, nigrostriatal and ceruleo-cortical, respectively, are significantly decreased in Parkinson’s disease (PD). As such, the five components of neurotransmission (biosynthesis, storage, release, reuptake, and post-synaptic function) have been studied for respective contributions to deficits in DA or NE signaling in PD. The range of approaches used to interrogate these pathways include defining PD-related genes and physiological regulation of catecholamine genes [
8,
9,
10,
11,
12], expression of catecholamine-regulating enzymes and transporters [
13,
14,
15,
16,
17,
18,
19,
20], post-translational modification of biosynthesis enzymes [
21,
22,
23,
24,
25,
26,
27,
28], neuron electrophysiological properties [
29,
30,
31,
32,
33,
34], release and uptake [
35,
36,
37,
38,
39,
40], pre- and post-synaptic receptor function [
22,
30,
34,
41,
42,
43,
44,
45], basal ganglia circuit function [
46,
47,
48,
49,
50,
51,
52], and growth factor signaling [
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64].
2. Insights of How Striatal DA Signaling Affects Locomotor Function Have Reached a Plateau
In the context of PD, DA is, by far, the most studied of the catecholamines, with NE running a distant second. Since 1962, there have been ~29,000 publications associated with DA and PD vs. ~1700 associated with NE and PD. The evidence for deficient nigrostriatal DA signaling as the primary cause of motor symptoms of PD is strong. Yet there still remains a critical unresolved issue that hampers progress: a continuous perseverating focus to attribute deficient DA signaling in the striatum as the sole culprit for motor impairment. This focus is undoubtedly driven by the longstanding working model of basal ganglia circuit dysfunction that arises from the loss of striatal DA due to the progressive loss of nigrostriatal neurons. It is argued that this striato-centric focus has generated a plateau in our understanding of exactly how any of the five steps of neurotransmission with deficient DA-regulating function in striatum actually impair motor function. For definition purposes, the relation of nigrostriatal DA signaling to motor impairment will focus upon bradykinesia/hypokinesia, which is among four cardinal signs of PD which also include rigidity and postural instability and tremor at rest. Indeed, there are clinically based examples of where improvements in striatal DA signaling did not equate to alleviation of motor impairment in PD patients [
60,
64,
65]. More evidence of this lack of alignment between striatal DA levels and severity of motor impairment is seen at the later stages of PD. Although the severity of motor impairment continues to worsen 4 to 5 years after PD diagnosis, loss of striatal DA-regulating proteins or signaling has already reached near 100% [
20,
66,
67,
68,
69]. There is a comparable amount of evidence for this misalignment between striatal DA levels and motor function status in pre-clinical studies of rat PD models [
22,
57,
58,
59,
70,
71,
72,
73,
74,
75,
76]. Motor impairment may also be present with far less than 80%, if any, striatal DA loss [
54,
65,
70,
72] or, conversely, motor impairment may not be present even though striatal DA loss meets or exceeds 80% [
22,
73,
74]. Motor impairment can also be alleviated without any increase in or recovery of striatal DA or DA-regulating protein loss [
54,
57,
59,
73,
74,
75,
76].
The weight of evidence that shows the influence of striatal DA signaling on basal ganglia circuits is too great to list here. However, the incongruities between the level of locomotor function and DA signaling in striatum can no longer be ignored if we are to solve which critical dopaminergic element(s) are to be targeted to maximize effective therapeutic strategies. The key question is where in the nigrostriatal pathway does DA have the greatest influence on locomotor function; particularly regarding the mechanisms that drive the initiation of self-generated movement. Although the evidence that nigral DA signaling can influence motor function is sparse, it has nonetheless been in existence since the 1980s [
77,
78,
79,
80,
81]. The paucity of studies evaluating the SN is likely due to a prevailing presumption that neurotransmitter functions at the axon terminal are the sole influence of behavioral outcomes. Thus, interrogation of nigral DA signaling has not been considered in experimental designs to define how components of nigrostriatal DA signaling affect locomotor activity. In this light, it is reasonable to presume that the numerous ambiguities between striatal DA regulation and motor function that have accumulated in the literature over the past several decades could have been resolved if assessment of nigral DA signaling was included in the study design.
3. Dissecting the Impact of the Five Components of DA Neurotransmission on Locomotor Function
As goes with the loss of nigrostriatal neurons in PD, the loss of DA-regulating proteins and processes involved in neurotransmission follows. Interference with the functions of any of these proteins or processes can also affect locomotor function in naïve (non-PD) animal models. Tyrosine hydroxylase (TH) is the rate-limiting step of DA biosynthesis, converting tyrosine to L-dihydroxyphenylalanine (L-DOPA). Inhibition of TH with alpha-methyl-p-tyrosine (AMPT) decreases DA tissue levels and inhibits locomotor activity [
7,
82,
83,
84,
85,
86]. In humans, inhibition of hyperkinetic movements, such as chorea, dystonia, or dyskinesia, can also be produced by AMPT [
87,
88]. The storage of DA and NE is controlled by vesicular monoamine transporter 2 (VMAT2), which imports monoamines like DA into synaptic vesicles using a proton gradient. This function is inhibited by reserpine, which also inhibits locomotor activity [
89,
90,
91], as first identified by a parkinsonian symptom side effect produced in hypertension treatment [
92]. VMAT2 is expressed in both striatum and SN [
93,
94], which confers the capacity for storing DA for eventual release in the entire nigrostriatal pathway.
Once DA is packaged in synaptic vesicles, it can be released by neuronal activity or by modulation of transporter function through stimulant action. At the extracellular level, DA release from the nigrostriatal pathway is the step that delivers tissue content, via vesicular delivery, to the synapse [
95,
96,
97,
98], wherein DA has four fates, binding to the pre- or post-synaptic DA receptors, reuptake into the neuron, or diffusion away from the release site [
99]. Drugs that target DA receptors, the post-synaptic DA D
1 receptor or pre- and post-synaptic DA D
2 receptor, also influence locomotor activity and are targets for pharmacotherapy in PD treatment [
100]. An acute regimen of antipsychotics such as haloperidol or either DA D
1 or D
2 receptor antagonists reduce locomotor activity [
101,
102,
103,
104,
105]. Conversely, DA D
1 or D
2 agonists increase locomotor activity in rodents and primates [
43,
106,
107] and improve motor functions in late-stage human PD [
108,
109,
110]. The release of DA can also be modulated by DA D
2 autoreceptor function [
111] in both striatum and SN [
31,
112]. Finally, it should be mentioned that although the focus of this review on DA receptors is upon the D
1 receptor, with brief overview of the D
2 receptor, the three other DA receptors have been recently shown to play a role in locomotor impairments of PD, particularly the D
3 and D
5 receptors [
113,
114,
115,
116].
Functionally, the regulation of DA release by neuronal activity is critical for initiation of locomotor activity [
117,
118,
119,
120,
121]. Deficits in DA release, such as occurs in aging or from over-expression of alpha-synuclein, are associated with decreased locomotor activity [
122,
123,
124]. Conversely, under conditions that increase DA release, such as induced by amphetamine or methamphetamine [
125,
126,
127], there is increased locomotor activity [
128,
129,
130].
The termination of DA signaling occurs by reduction of extracellular DA levels in the synapse, largely, though not exclusively [
99], through reuptake by the dopamine transporter (DAT) [
131,
132,
133]; a process that occurs in SN as well as striatum [
134,
135,
136]. DAT protein expression is considerably greater in the striatum [
94], and, not withstanding possible influences of trafficking or contributions of other monoamine transporters, this difference may explain why DA release and uptake dynamics differ between these two regions [
134,
135,
136]. Through constant trafficking between cytosol and plasma membrane, DAT function is dynamically regulated, including aging and in PD [
137,
138,
139]. The DAT, like the DA D
2 receptor, also has considerable interaction with other components of DA neurotransmission, including DA D
2 receptors [
34,
112], and has considerable influence on maintaining DA tissue levels, TH expression, and phosphorylation selectively in the striatum, but not in SN [
140,
141]. There is also evidence of plasticity in DA uptake under conditions where DA and DAT levels are particularly low. In such cases, the NE transporter may also transport DA, with inherently low DA innervation or from severe loss of nigrostriatal neuron terminals [
142,
143].
Given the considerable influence of DAT on DA homeostasis, locomotor activity is strongly affected by DAT expression levels. DAT knockout mice or rats show a hyperkinetic phenotype [
144,
145,
146]. This hyperkinetic phenotype is not likely explained by the low DA uptake capacity in the striatum due to DAT knockout, as DA tissue content levels are severely reduced to a level that is comparable to nigrostriatal lesion (>90% loss) [
140,
141]. Systemic delivery of nomifensine, a DAT inhibitor, increases locomotor activity [
147], consistent with the hyperkinetic phenotype of the knockout [
144,
145,
146]. While presumably this effect would be considered to be due to elevated extracellular DA levels in striatum from interference with DA uptake, we recently reported that infusion of nomifensine in striatum did not increase locomotor activity in aged rats, despite a striatum-specific increase in extracellular DA levels produced by nomifensine infusion therein [
148].
4. Nigrostriatal DA Signaling and PD-Related Motor Impairment
From the perspective of deficient DA signaling impact on motor impairment in PD, a long-standing unresolved issue is why motor impairment does not occur until there is 70–80% TH or DA loss in striatum. It was long thought that increased DA turnover reflected increased DA signaling during progressive loss of the nigrostriatal neuron terminals [
19,
73,
172,
173,
174,
175], thus compensating for TH protein loss to enable normal locomotor activity. L-DOPA, the product of TH, remains the gold standard for treating motor symptoms. Thus, it stands to reason that compensating for TH loss through engagement of innate compensatory mechanisms that increase DA levels would promote maintaining locomotor function until striatal TH loss was too severe.
Increased DA turnover was proposed to be an indicator of enhanced DA signaling to compensate for TH protein loss during nigrostriatal neuron loss [
19,
52,
73,
172,
173,
174,
175]. However, Bezard and colleagues definitively showed in an elegant timeline study using MPTP-lesioned primates in which increased DA turnover occurred only after bradykinesia manifested; there was no evidence of increased DA turnover during the asymptomatic period [
19]. Also, 80% TH and DA loss in striatum appeared to be necessary for the onset of bradykinesia; even 60% TH loss in striatum was observed during the asymptomatic period. Fortunately, this study also assessed TH loss in the SN and found that, at the onset of motor impairment, there was ~40% loss in the SN; far less than 80% loss seen at the axon terminals. This loss in the SN may be related to regionally selective loss of nigral neurons, as shown in human aging and PD [
159,
161]. Also, this disparity in TH loss between SN and striatum has strong translational relevance because this disparity consistently manifests in human PD [
20,
66,
67,
68]. Nonetheless, the lack of evidence to support a role for increased DA turnover in striatum to offset the onset of locomotor impairment gave rise to the consideration that non-DA related mechanisms to be responsible for delaying the onset of motor impairment [
52].
Recent work from our group indicates that the compensatory mechanism to mitigate the severity of hypokinesia and delay its onset against progressive nigrostriatal neuron loss is related to increased DA signaling in the SN, and not striatum [
22]. This mechanism involves an increase in ser31 TH phosphorylation, specifically in the SN, that begins early after nigrostriatal loss induction by 6-hydroxydopamine (6-OHDA) and is maintained at least until neuronal loss reaches 80% in the SN. As a result of this increase in ser31 TH phosphorylation, there is less loss of DA as compared to TH throughout neuronal loss [
22]. This differential in DA and TH loss also manifests in the SN, contralateral to the lesioned side, as TH loss begins there at a later time after lesion induction. When correlating the loss of DA in SN and striatum against the severity of motor decline in the open field, only DA loss in the SN has significant correlation [
22]. In striatum, we found no difference in TH and DA loss, as both exceeded 90% early after lesion induction, commensurate with decreased ser31 TH phosphorylation and increased DA turnover. In contrast, DA turnover decreased in the SN as neuron loss progressed. Our findings of diminished lesion impact on DA tissue content in the SN are also reflected in the extracellular realm, wherein baseline DA levels are unaffected by 6-OHDA lesions despite severe neuronal loss [
176]. Together, these results frame a new perspective on the mechanism by which motor impairment is delayed by increased DA biosynthesis in the SN, despite progressive nigrostriatal neuron loss that occurs in PD (
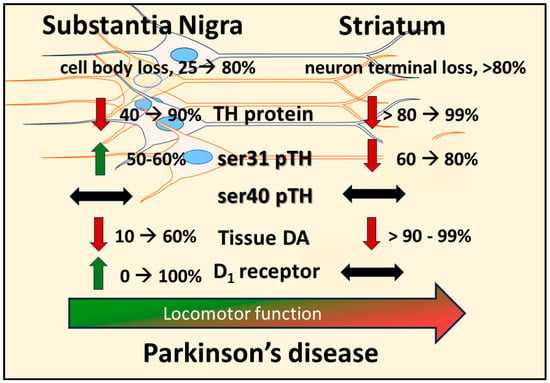
Figure 2). Moreover, these results are disease-relevant and further support a role for nigral DA signaling in locomotor function.
Figure 2. Disparate molecular changes in dopamine (DA) signaling components in substantia nigra (SN) and striatum in response to neuronal loss and relation to decreased locomotor function. Induction of nigrostriatal neuron loss by 6-OHDA produces a progressive loss of neurons over 4 weeks. Loss of tyrosine hydroxylase (TH) protein in SN is less than the magnitude of loss in the striatum at the earlier time points post-lesion, and tissue DA loss is substantially and consistently less in the SN than in striatum. In response to TH loss, there is a site-specific increase in TH phosphorylation at ser31 (ser31 pTH), not ser40 (ser40 pTH), restricted to the SN; whereas in striatum, there is a progressive decrease in ser31 pTH. This increase in ser31 pTH in the SN offsets the progressive loss of TH therein to keep DA loss at a lower level than TH. As DA tissue loss increases in the SN, the DA D1 receptor (D1) increases expression at the latter stages of neuron loss. The increases in both ser31 TH phosphorylation and D1 in the SN are compensatory mechanisms to delay the onset of locomotor impairment and alleviate its severity.