Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Medicinal
Microorganisms participating in the development of biofilms exhibit heightened resistance to antibiotic treatment, therefore infections involving biofilms have become a problem in recent years as they are more difficult to treat. Consequently, research efforts are directed towards identifying novel molecules that not only possess antimicrobial properties but also demonstrate efficacy against biofilms. While numerous investigations have focused on antimicrobial capabilities of Schiff bases, their potential as antibiofilm agents remains largely unexplored.
- Schiff base
- antibiofilm
- antimicrobial
- imines
1. Introduction
Clinically relevant microbial biofilms are defined as “aggregated microbial cells surrounded by a polymeric self-produced matrix, which may contain host components”, suspended or attached to a surface [1]. Biofilm-related infections attracted the attention of scientists 50 years ago in the context of cystic fibrosis, and their impact on the medical field has grown ever since [2]. These infections may be tissue-related (chronic otitis media, chronic sinusitis, chronic laryngitis, dental plaque, endocarditis, cystic fibrosis, kidney stones, biliary tract infections, urinary tract infections, osteomyelitis, wound infections, etc.) or associated with medical devices (contact lenses, endotracheal tubes, cardiac devices or catheters) [1,3]. Some examples of biofilm forming pathogens are: Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Staphylococcus epidermidis, Streptococci, Enterococci and Candida spp. [4].
Biofilm formation requires four stages: (i) attachment of the mobile microorganism to a surface, (ii) colonization, (iii) development and maturation of biofilm and (iv) dispersion and propagation [5,6]. Attachment is mediated by cilli, flagella, surface proteins of microorganisms and rugosity of the surface [7]. It is reversible at first and then becomes irreversible, triggering transcription of specific genes for signalling molecules and extracellular polymeric substances (EPS). Colonization involves growth and division processes and EPS synthesis [5]. A mature biofilm consists of three layers: the biofilm nucleus, membranes of basal microorganisms and external mobile planktonic cells. It is a complex mixture of water, microbial cells, proteins, aminoacids and polysaccharides [6]. Dispersion is mediated by external factors or by self-digestion and contributes to dissemination of infection [7]. There are two important features of biofilm (sessile) growth compared with the free-floating (planktonic) state that contributes to pathogenicity: increased tolerance to antibiotic treatment and persistence in the host, despite inflammation and immune response [1]. The major consequence is that biofilm infections are hard to treat and usually become chronic [8].
Antibiotherapy is active on planktonic microbial cells, but its effectiveness against sessile states is variable, as established biofilms are usually recalcitrant to conventional antibiotics [9]. Treatment may require higher doses of antimicrobials, prolonged duration [8,10], combination therapy [11,12] or special modes of administration (nebulized antibiotics) [13].
There is a constant need to develop alternative antibiofilm strategies, and extensive research has been conducted in this direction [10]. Antibiofilm small molecules are relevant because they target stages in biofilm development which are different to those of normal planktonic state [9]. The mechanisms (Figure 1) may involve: blocking microbial adhesion (biocides [14], antibiotics [15] and impregnated coatings [16]), inhibition of microbial communication (quorum sensing inhibitors and quorum quenching) [17] and killing cells inside the biofilm (persisters or non-growing cells) (cisplatin, cis-2-decenoic acid, colistin, mytomicin C) [18,19,20,21]. In particular, strategies like quorum sensing inhibition may prove useful because they do not necessarily affect bacterial growth but they reduce virulence, thus increasing susceptibility of microorganisms to antibiotics and to host immune cells without the risk of antibiotic resistant [17].
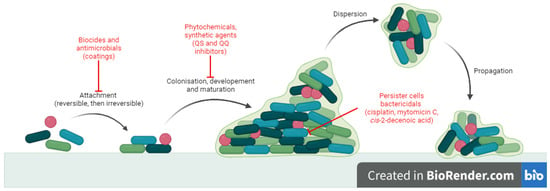
Figure 1. Antibiofilm mechanisms of action for small molecules.
Schiff bases are compounds with the structure R’N=CR2 (R’ ≠ H) [22], traditionally formed in the reaction of alkyl/aryl aldehydes or ketones with primary amines [23]. Many considered them to be synonymous with azomethines (RN=CR2, R ≠ H), both being a particular case of imines (RN=CR2, R = H, hydrocarbonyl) [22]. They are all part of carbonyl compound derivatives, formed in the reaction with basic nucleophiles (amines and their derivatives—hydroxylamine, hydrazine, N-acyl-hydrazine and semicarbazide). Therefore, this class of compounds also includes oximes (RR’C=NOH), hydrazones (R1R2C=N-NH2), N-acyl-hydrazones (R1R2C=N-NH-CO-R) and semicarbazones (R1R2C=NNH-(CO)-NH2) [14].
Schiff bases have numerous applications including coordination chemistry [23], catalysis [24], chemosensors [25] and intermediates in synthesis [26,27]. They also exhibit a variety of biological applications: antibacterial [28,29,30,31,32,33,34], antifungal [34,35,36], antiviral [37,38], antimalarial [39,40], antituberculosis [41,42,43], anthelmintic [44,45], urease inhibitors [46,47,48], anticancer [49,50,51], antidyslipidemic [52], antidiabetic [53,54], antidepressant [55,56,57], anticonvulsant [58,59], neurodegenerative disorder treatment [60,61], anti-inflammatory [62] and antioxidant [53,54].
Human and veterinary therapy benefits from several antibacterial drugs recognized as Schiff bases. In multidrug-resistant tuberculosis (MDR-TB), a longer treatment regimen includes two Schiff bases which act on Mycobacterium tuberculosis cell wall: bacteriostatic terizidone (Figure 2), a cycloserine derivative, analogue of D-alanine and anti-leprosy clofazimine, which is an iminophenazine [63,64].
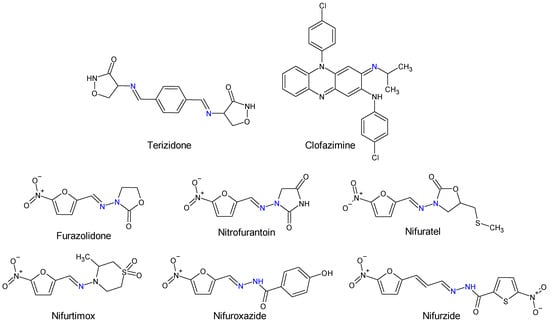
Figure 2. Structure of Schiff base and N-acyl-hydrazone medicines.
Oximes and hydrazones are moieties frequently used in medicinal chemistry. Examples of oxime drugs utilized in antimicrobial therapy include: cephalosporins (second-generation—cefuroxime, third-generation—cefdinir, cefixime, cefpodoxime, ceftazidime, cefmenoxime, ceftizoxime, ceftriaxone, cefotaxime, cefpirome, fourth-generation—cefepime, fifth-generation—ceftaroline, cefiderocol) [65,66,67,68], as well as antifungal (oxiconazole) [69], antiviral (enviroxime, zinviroxime) [70,71] and anti-infective (nifuroxime) medications [72] (Figure 2 and Figure 3).
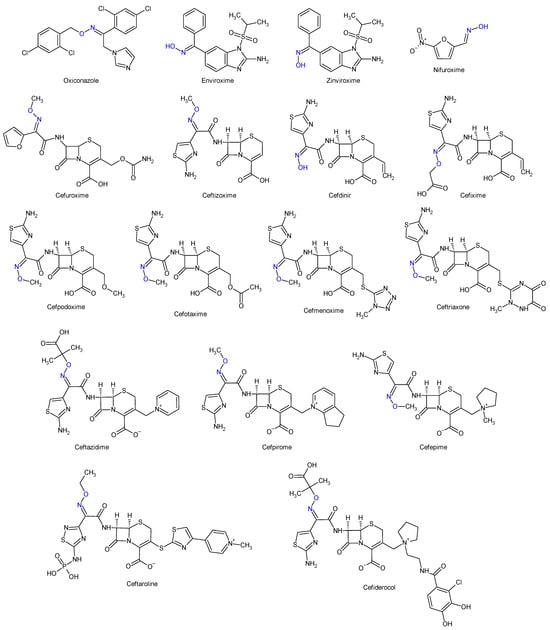
Figure 3. Structure of oxime medicines.
N-acyl-hydrazones derivatives of 5-nitrofuran are prodrugs which act against different types of pathogens [73]. Some examples are nifuratel (antibacterial, antifungal, antitrichomonal agent) [74], furazolidone (antiprotozoal agent, gynaecological antiinfective and antiseptic) [75,76], nifurzide, nifuroxazide (intestinal antiinfectives, antidiarrheal agents) [77], nitrofurantoin (antibacterial) [78] and nifurtimox (antitrypanosomal and antileshmanial agent) [79]. Along with their stated antiinfective indications, studies have explored other possible applications of these drugs. Nifuratel—activity against Leishmania spp. [80,81], nifuroxazide—quorum sensing and biofilm inhibition [82], antischistosomal activity [83]. N-acyl-hydrazones 5-nitrofurans also exhibit anticancer properties, inhibiting different pathways in cancer cell cycles: signal transducer and activator of transcription 3 (STAT3) (nifuroxazide [84], nifuratel [85]), aldehyde dehydrogenase 1 (ALDH1) (nifuroxazide [86]) and nuclear factor kappa B signalling (furazolidone [87]) (Figure 2 and Figure 3).
2. A Possible Strategy to Combat Biofilm-Related Infections
Due to their ease of synthesis and their wide range of applications, salicylaldehyde Schiff bases are frequently cited in the relevant literature. These compounds demonstrate antimicrobial potential, both as simple ligands and as metal complexes [56,57,58]. The antimicrobial activity is directly influenced by substitutions on the salicyl moiety, with halogenation exerting a noticeable impact in particular [58].
Taurine-5-bromosalicylaldehyde Schiff base (TBSSB) is a potassium salt of 2-{[1-(5-bromo-2-hydroxyphenyl)-meth-(Z)-ylidene]-amino} ethanesulfonic acid, with antistaphylococcal [97] and antimycobacterial potential [98] that is active against both planktonic and sessile forms. TBSSB was bactericidal against S. aureus (MIC 32 μg/mL), affecting membrane integrity and also preventing biofilm formation at 8 μg/mL [97]. The sulfonic acid group seems essential to antistaphylococcal activity [97]. The antimycobacterial effect was even better. TBSSB completely inhibited M. smegmatis mc2155 growth at > 60 μg/mL, presenting greater cell wall destruction compared with S. aureus alongside alterations in cell division. Additionally, it exhibited dose-dependent inhibition of Mycobacterium biofilm formation [31].
p-Aminobenzoic acid (PABA) is an amino acid derivative, implicated in folate biosynthesis in microbial cells [133]. Due to its importance for bacterial viability, it serves as a target for antimicrobial therapy [134]. Therefore, obtaining hybrid molecules is a direction of molecular development [135] in the search for new anti-infective agents.
Starting from a series of Schiff base derivatives of p-aminobenzoic acid and halogenated salicylaldehydes (compound 1), me-too analogues were synthesized and tested for antimicrobial, antibiofilm and cytotoxicity activities [99]. The design approaches were as follows: isomerization (m-aminobenzoic acid (MABA) derivatives 2) esterification (methyl esters 3, and ethyl esters 4) amide formation (N-phenylamides 5) duplication of azomethine bond (3,5-diaminobenzoic acid (DABA) derivatives 6). The Schiff bases obtained were active against Gram-positive strains, having MIC from 7.81 μM. The corresponding amines presented no antimicrobial effect. Diiodo derivatives (2b, 3b and 6b) were comparable in action to bacitracin (SA: MIC 7.81 μM, EF: 15.62 μM). No activity was observed against Mycobacterium strains. Regarding antifungal activity, the analogues surpassed the original PABA Schiff bases. Derivatives 2, 5 and 6 exhibited broad-spectrum activity, C. albicans and T. interdigitale being the most susceptible. The best results were obtained for diiodo analogues (2b, 5b, 6b), having MICs comparable to fluconazole (CA: 6.5 μM). The antibiofilm evaluation was performed on two strong biofilm producers: methicillin-resistant S. aureus ATCC 43300 and S. epidermidis ATCC 1228. Compound 3b (methyl (E)-4-[(2-hydroxy-3,5-diiodobenzylidene)amino]benzoate) was only moderately active (MRSA: MBIC 781.25–1562.5 μg/mL, MBEC 1562.5–3125.0 μg/mL; SE: MBIC 781.25–1562.5 μg/mL, MBEC > 1562.5 μg/mL) compared with ciprofloxacin (MRSA: MBIC 0.381 μM, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.7625 μg/mL, MBEC 97.6–195.3 μg/mL). The methyl ester was also the least cytotoxic. Thus, 3,5-dihalogenosalicylic scaffold is essential for antimicrobial activity—iodine atoms preferred (3,5-diiodo, followed by 3-iod-5-chloro- substitution) [99].
Simplifying the structure of rafoxanide (a veterinary anthelmintic) by changing the amide group with azomethine and eliminating the phenoxy substituent, an imine analogue, (E)-2-{[(4-chlorobenzyl)imino]methyl}-4,6-diiodophenol (7) was obtained [100]. Compound 7 presented selectivity on Gram-positive bacteria, exhibiting antistaphylococcal (MIC 15.625–62.5 μM) and antienterococcal (MIC 62.5–125 μM) activities on reference strains and clinical isolates. The action is bactericidal, and the mechanism indicated inhibition of protein synthesis pathways followed by inhibition of nucleic acid and peptidoglycan production. It exhibits moderate-to-good antibiofilm activity against MRSA and SE (MRSA: MBIC 62.216–124.432 μg/mL, MBEC 124.432–248.863 μg/mL; SE: MBIC 31.108–62.216 μg/mL, MBEC 124.432–248.863 μg/mL) compared with ciprofloxacin (MRSA: MBIC 0.381 μM, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.763 μg/mL, MBEC 97.6–195.3 μg/mL). Due to its bactericidal action, compound 7 seemed to reduce bacterial metabolic activity and inhibit the viability of the released planktonic cells from the biofilm [100].
Combining two pharmacophores—salicylaldehyde and sulphonamides—two series of Schiff base analogues of sulfamethoxazole (compounds 8), sulfathiazole (compounds 9) and sulfamethazine (compound 10) were synthesized [101]. The influence of the substitution of salicylaldehyde moiety (R2) on antimicrobial activity was investigated. Gram-positive bacteria, especially Staphylococci, were susceptible to the action of the analogues (MIC ≥ 15.62 μM), including clinical isolates (MIC ≥ 3.91 μM) and resistant species (methicillin-resistant S. aureus, MRSA, cotrimoxazole resistant species). Interestingly, the Schiff bases were bactericidal in action compared with sulfonamides and active against cotrimoxazole resistant bacteria, exhibiting no cross-resistance. Eight compounds (8c–d, 9b–d, 10a–c) had MICs (15.62–31.25 μM) comparable to bacitracin (MIC 7.81-15.62 μM) against S. aureus. Once more, the most favourable outcome was achieved with the 3,5-dihalogen substitution on the salicylaldehyde molecule, particularly with the presence of at least one iodine atom, making sulfamethazine derivatives (10) the most potent [101].
4-[(3,5-Dichloro-2-hydroxybenzylidene)amino]-N-(4,6-dimethylpyrimidin-2-yl)benzene-sulfonamide (10a) inhibited MRSA and S. epidermidis biofilm formation (MBIC 390.6–781.25 μM, MBEC > 3462 μM) being inferior to ciprofloxacin (MRSA: MBIC 0.381 μg/mL, MBEC 48.8 μg/mL, SE: MBIC 0.381–0.763 μg/mL, MBEC 97.6–195.3 μg/mL). The compound was not able to disrupt the preformed matrix [101].
5-(4-Methylpiperazin-1-ylsulfonyl)benzylidene)anilines (11a–f) were synthesized and evaluated for antibacterial and anti-Candida actions [102]. The antibacterial activity varied among the strains and was influenced by the radical R used. B. subtilis was the most susceptible, followed by P. aeruginosa. Unsubstituted 11a was more potent than the reference (ciprofloxacin—MIC 50 μg/mL) against PA, with electron-donating groups (4-OCH3, 11f) increasing the activity. For S. aureus and E. coli biofilm inhibition, the compounds were inferior to ciprofloxacin. The most favourable substituent was the electron-withdrawing CF3 in ortho or in meta position (11b, 11c). Electron-donating OH seemed essential for antifungal and antibiofilm activity. Derivatives 11d (2-OH), 11c (3-CF3) and 11e (4-OH) surpassed fluconazole (MIC 50.0 μg/mL) in terms of anti-Candida activity. A similar trend was observed for fungal antibiofilm action, with compound 11d (2-OH) being the most active, followed by 11a (H), 11b (3-CF3) and 11e (4-OH) (fluconazole, IC50 40 μM). Compound 11d (2-(2-Ethoxy-5-(4-methylpiperazin-1-ylsulfonyl)benzylideneamino)phenol) inhibited the formation of C. albicans biofilm without affecting planktonic cells, which may indicate a quorum sensing mediated mechanism of action. The docking study against Candida secreted aspartyl protease (SAP5), the enzyme responsible for cell-to-cell adhesion and biofilm formation [136], indicated that the 11d is held in place by van der Waals interactions, while 4-methylpiperazine ring form hydrophobic interactions with amino acids at the active site. The azomethine group is also responsible for strong van der Waals hydrophobic and charge bonds interactions with important active site amino acid residues (Ile12, Lys83, Gly85, Asp86, Gly220, Thr221, Thr222, Thr222, Ile223, Tyr225 and Ile305) [102].
4-(o-Methoxyphenyl)-2-aminothiazole was reported to possess antibacterial and antibiofilm potential [137]. Its Schiff bases with substituted salicylaldehydes (12a–f) and 2-hydroxy-1-naphtylaldehyde (12g) were synthesized and evaluated for the same effects [103].
4-Bromo-2-(((4-(2-methoxyphenyl)thiazol-2-yl)imino)methyl)phenol (12f) and 2-(((4-(2-methoxyphenyl)thiazol-2-yl)imino)methyl)naphthalen-1-ol (12g) exhibited antibacterial action against B. subtilis (MIC 25 μg/mL). Compound 12g was also active against E. coli (MIC 100 μg/mL) [103], with Schiff bases surpassing the parent amine (amine: MIC 250 μg/mL for B. subtilis, 500 μg/mL for E. coli) [137]. Regarding antibiofilm potential, compounds 12f and 12g were able to inhibit P. aeruginosa biofilm formation but they do not affect the viability of the cell, suggesting a quorum sensing mechanism of action [103].
A series of Schiff bases starting from 2-amino-5-chloro-benzophenone was obtained using microwave irradiation and evaluated for antibiofilm and antibacterial activity [104]. Twelve compounds presented MBIC under 100 μg/mL (13a–k). The antibacterial/ antibiofilm activity depended on the type and nature of substituents (R, R1), with electron-donating groups (methoxy, hydroxy) and halogens being favourable. The salicylaldehyde derivative (13d) was only active against S. mutans. The introduction of halogen atoms extended the action to S. aureus (F—13e), K. pneumoniae (Br—13f) and P. mirabilis (Cl, Br—13g). The acridine derivative (13l) and compounds 13a–c inhibited both Gram-positive and Gram-negative bacteria, being inferior to cefixime (MIC 41 μg/mL). S. aureus biofilm was significantly disrupted by compounds 13i, 13k and 13g, while 13i was also active against preformed biofilm of P. mirabilis [104].
4-aminophenazone Schiff bases with different substituted cinnamaldehydes (14a–c) were obtained and tested for antimicrobial and antibiofilm activity. 4-(2-Bromo-3-phenyl-2-propenylideneamino)-1,5-dimethyl-2-phenylpyrazol-3-one (14a) exhibited broad antimicrobial spectrum. It inhibited all fungal strains and all tested bacteria, except P. aeruginosa, exhibiting bactericidal (K. ozaenae, S. enterica) or bacteriostatic effect (E. gergoviae). It reduced up to 90.41% of the biofilm of C. tropicalis and between 75–83% of E. faecalis and S. aureus. Compounds 14b and 14c were also active on biofilm [105].
Bacterial fatty acid synthetase may serve as the target for the development of new antibacterial agents. Triclosan and other 2-hydroxydiphenyl ethers demonstrated inhibition against enoyl-acyl carrier protein reductase (FabI), a key enzyme in fatty acid production [138,139]. Schiff bases and hydrazones have also been reported as inhibitors of staphylococcal β-ketoacyl carrier proteinsynthase III (encoded by FabH gene) [140,141]. In Gram-negative bacteria, PqsD—an enzyme implicated in Pseudomonas autoinductors synthesis—is structurally related to FabH. Thus, inhibitors of fatty acid synthetases may also act against PqsD [142].
Linezolid derived Schiff bases were synthesized starting from 4-(4-amino-2-fluorophenyl)-morpholine in order to obtain PqsD enzyme inhibitors [106]. Biofilm inhibition varied according to radical R used. The quinoline derivatives (N-((2-chloroquinolin-3-yl)methylene)-3-fluoro-4-morpholinoaniline—15h, N-((2-chloro-8-methylquinolin-3-yl)methylene)-3-fluoro-4-morpholinoaniline—15i) exhibited the greatest activity against P. aeruginosa biofilm (15h—IC50 12.97 ± 0.33 μM,15i—IC50 15.63 ± 0.20 μM), surpassing linezolid (IC50 15.93 ± 0.18 μM). They also presented good anti-Pseudomonas activity, comparable to linezolid (MIC 2.5 μg/mL) and antimicrobial action against E. coli and B. subtilis (15h, 15i: MIC 5–10 μg/mL, linezolid: MIC 2–3 μg/mL). The docking studies of 15h and 15i against PqsD enzyme revealed van der Waals interactions, hydrophobic bonds (methylene groups of morpholine, methyl group of quinoline) and hydrogen bonds (fluorine atom of linezolid moiety, azomethine group) with amino acid residues of the active site (Figure 4). The phenyl derivatives (15a–e) exhibited only moderate antibiofilm and antimicrobial activities, with electron-donating groups (halogen and methoxy) slightly increasing the effect. Indolyl (15g) and furanyl (15f) derivatives presented no enhancement (Figure 4) [106].
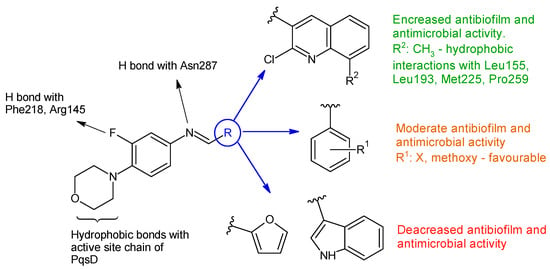
Figure 4. Structure–activity relationship for compounds 15, including interactions with PqsD enzyme.
N-(3-(-2-(7-chloroquinolin-2-yl)vinyl)benzylidene)anilines 16a–j were synthesized based on 2-n-heptyl-4-hydroxyquinoline (HHQ) and 2-n-heptyl-3-hydroxy-4(1H)-quinoline (PQS- Pseudomonas Quinoline Signal) [107]. Radical R on the phenyl ring influenced antimicrobial and antibiofilm activities. Electronegative substituents improved antibacterial activity, with the p-chloro derivative 16b being more potent than ciprofloxacin (MIC 50 μg/mL) on E. coli, while the p-trifluoromethyl derivative 16g was the only one active on P. aeruginosa (MIC 91.5 μg/mL). S. aureus was most sensitive to o-trifluoromethyl derivative 16e (MIC 55.3 μg/mL). For antifungal activity, the p-chloro substitution was beneficial, followed by unsubstituted derivative, while the p-trifluoromethyl derivative presented lower activity (16f: IC50 174.4 μg/mL). Compounds with electronegative substituents such as trifluoromethyl (N-(3-(2-(7-Chloroquinolin-2-yl)vinyl)benzylidene)-4-(trifluoromethyl)aniline—16g, or chloro (N-(3-(2-(7-chloroquinolin-2-yl)vinyl)benzylidene)-4-chloroaniline—16b) on para position presented good fungal antibiofilm activity, comparable to fluconazole (IC50 40 μM), whereas methoxy (16h) and nitro (16i, 16j) derivatives indicated moderate activity (IC50 < 100 μM). The lack of substitution or hydrophobic groups (CH3) was unfavourable for biofilm inhibition (Figure 5). Docking studies against agglutinin-like protein (C. albicans Als-3 adhesin) indicated the formation of halogen bonds between para electronegative substituents and the active site, hydrogen bonds between imine group and Tyr21 and Tyr226 and π-π stacking interaction between naphthyl ring and Leu293, Val161, Trp295, Tyr166 and Val172 (Figure 5) [107].
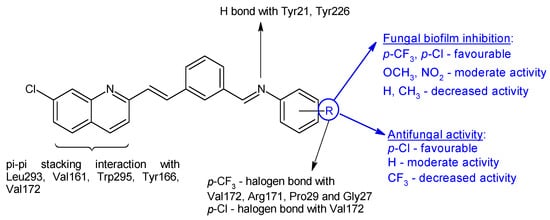
Figure 5. Structure–activity relationship for compounds 16, including interactions with Als-3 adhesin of C. albicans.
N-phenyl-3-cyano-4-amino-pyrazole was used as the starting point for the design and development of antifungal Schiff bases. For both antifungal and antibiofilm activity, the optimal substituents were electron-withdrawing Br, NO2 and COOH. Compound 17i (5-(4-bromobenzylideneamino)-1-(2,6-dichloro-4-(trifluoromethyl)phenyl)-1H-pyrazole-3-carbonitrile) was the most potent(MIC 42.6 μg/mL, IC50 41.5 μM), and is comparable to fluconazole (MIC 50 μg/mL, IC50 40 μM) [108].
P. aeruginosa uses two QS systems, las and rhl, that rely on transcriptional activators (LasR and RhlR, respectively) and autoinducer molecules (N-3-oxo-dodecanoyl-L-homoserine lactone, respectively N-butyryl-L-homoserine lactone) [143]. Interfering with these systems may serve as a strategy to reduce virulence and pathogenicity [144].
Combining 6-amino-4-(thiophen-2-yl)-2-oxo-pyridine-3,5-dicarbonitrile (18) and 6-amino-4-(furan-2-yl)-2-oxo-pyridine-3,5-dicarbonitrile (19) with aromatic aldehydes and ketones, two series of antimicrobial Schiff bases (18a–c and 19a–c) were synthesized under both conventional and green conditions (ceric ammonium nitrate catalysis) [109]. Biological screening revealed significant antimicrobial potential for azomethines, especially on Gram-negatives, surpassing or equalizing references (gentamicin and fluconazole) and biocidal modes of action. In terms of biofilm inhibition, azomethines were active against all tested strains, with MRSA and E. coli biofilms being the most susceptible. They were able to reduce LasR gene expression with 10–40% at 1/8 MIC compared with 60% for doxycycline. Compound 19a presented extended antibacterial spectrum (E. coli- MIC 125 μg/mL, K. pneumoniae- MIC 15.6 μg/mL) comparable to gentamicin (MIC 250, 250 μg/mL). Compound 19b was the most active against MRSA (MIC 62.5 μg/mL, gentamicin—MIC 125 μg/mL), with 5-bromo substituent on the phenyl ring being essential. Compound 18c exhibited antifungal properties, surpassing fluconazole (MIC 62.5 μg/mL) and other derivatives (MIC 250 μg/mL), however, its effect on fungal biofilm was reduced (15.15%). Derivative 19c (6-Amino-1-((1,3-dioxo-1,3-dihydro-2H-inden-2-ylidene)amino)-4-(furan-2-yl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile) was active against P. aeruginosa biofilm in both planktonic and sessile forms (gentamicin—BI 29.4%). It presented the highest degree of LasR gene expression inhibition among tested compounds (40%), significantly reduced C. albicans biofilm (75.0%) and surpassed fluconazole (57.6%). Structure–activity relationships revealed that azomethine is important for activity, which varied according to the substituents in position one (imine groups) and four (furanyl, thiophenyl) on pyridine moiety (Figure 6). Benzylidene and 1,3-dioxo-1,3-dihydro-2H-inden-2-ylidene increased the antibacterial spectrum, with electron-donating groups (hydroxy, ethoxy, bromo) being beneficial for MRSA and E. coli biofilm inhibition [109].
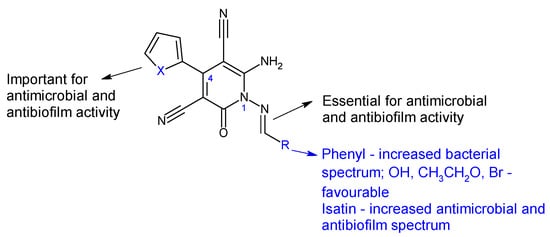
Figure 6. Structure–activity relationship for compounds 18, 19.
4-Amino-3-mercapto-6-(trifluoromethyl)-1,2,4-triazin-5(4H)-one was used as the starting point for the synthesis of six Schiff bases (20a–f) [110]. The in vitro biological evaluation revealed that Gram-positive and S. typhi were susceptible to all azomethines, while E. coli was tolerant to 20a and 20e. Halogen-substituted compounds 20b–d exhibited broad spectrum antibacterial action, inhibiting in different percentages all tested strains (IR 4.57–87%). Compound 20b (4-((4-fluorobenzylidene)amino)-3-mercapto-6-(trifluoromethyl)-1,2,4-triazin-5(4H)-one) was the most potent against E. coli and S. aureus (IR 87%, 75%; MIC 3.90 μg/mL) (ciprofloxacin—MIC 0.39 μg/mL). Compound 20a (4-(ethylideneamino)-3-mercapto-6-(trifluoromethyl)-1,2,4-triazin-5(4H)-one) was most active against S. typhi, with a MIC of 7.81 μg/mL. Regarding antifungal assay, A. flavus was more sensitive to the action of Schiff bases than A. niger. Compound 20a displayed good antifungal activity (IR up to 87%, MIC 15.62 μg/mL for A. flavus). Compounds 20c and 20f exhibited moderate fungal inhibition (IR 43–82%) but had better MICs than nystatin (3.90 μg/mL compared with 8.25 μg/mL for A. flavus). Phenyl derivatives 20b–f inhibited the biofilm of E. coli and S. aureus, with 20b being the most potent (IR 87.4%—E. coli, 72.4%—S. aureus) while 20a was inactive. Hence, fluoro in para position of the benzene ring (20b) improves antibacterial and antibiofilm activities, whereas chloro and trifluoromethyl groups are beneficial for antifungal action (Figure 7) [110].
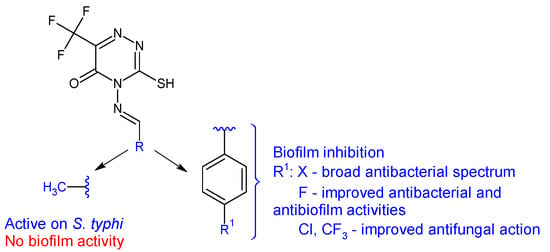
Figure 7. Structure–activity relationship for compounds 20.
Isatins (1H-indole-2,3-diones) are synthetic and also naturally-occurring compounds, largely employed in organic synthesis due to their versatility and numerous applications [145].
A series of hybrid Schiff bases (21a–f) were synthesized by incorporating isatin, pyrazole and either piperidin-1ylsulfonyl or N-methylpiperazin-1ylsulfonyl into a single molecule [111]. These resultant molecules are amphiphilic in nature, stemming from the combination of polar groups (NH2, OH, SO2) with lipophilic hydrocarbon components, thereby enhancing their antibacterial potential. Compounds 21a–d exhibited good antibacterial activity against tested strains (MIC 53.45–258.32 μM), comparable to norfloxacin (MIC 100.31–200.63 μM) and ciprofloxacin (MIC 48.33–96.68 μM). 5-Aminopyrazole moiety (R2: NH2) performed better than 5-hydroxypyrazole (R1: OH), with the most potent derivatives being 21b and 21d. Compounds 21b–f exhibited good antifungal activity (MIC 106.91-208.59 μM), surpassing fluconazole (MIC 220.76 μM). Derivatives 21a–d were fungicidal and bactericidal against all strains, except S. aureus (21b—bacteriostatic), E. coli (21a—bacteriostatic). Compound 21d ((E)-3-({5-amino-1-benzoyl-4-[(E)-(4-hydroxyphenyl)diazinyl]-1H-pyrazol-3-yl}imino)-5-(piperidin-1-ylsulfonyl)indolin-2-one), the most active antimicrobial, inhibited the MRSA biofilm formation at concentrations of 0.007–0.03 mg/mL (BI 70.8 ± 2.3–89.9 ± 4.7%) [111].
In a subsequent study, Schiff bases and other imine derivatives were obtained condensing 5-((4-methyl-piperazin-1-yl)sulfonyl)indoline-2,3-dione with aminothiazole derivatives, sulfathiazole or thiourea [112]. Compounds 22a–c exhibited good antibacterial activity (MIC 1.9–125 μg/mL), outperforming levofloxacin (MIC 8.1–130 μg/mL) with compound 22c being the most active. Antifungal potential was reduced and compounds 22a and 22d presented moderate activity (MIC 62.5, 31.2 μg/mL), respectively (nystatin—MIC 3.9 μg/mL). Compounds 22a–d were active against S. aureus biofilm (BI50 1.95–15.6 μg/mL), with the sulfathiazole derivative 22b (4-((5-((4-Methylpiperazin-1-yl)sulfonyl)-2-oxoindolin-3-ylidene)amino)-N-(thiazol-2-yl)benzenesulfonamide) being the most potent. Compounds 22c, 22d and 22e were also active in this order against P. aeruginosa biofilms (BI50 7.8 ± 0.13, 15.6 ± 0.32, 31.25 ± 0.051 μg/mL). Derivatives 22a–c, especially 22c, inhibited QS system of E. coli, known as fsr, thus presenting a QS mechanism of antibiofilm action [112].
Starting from two modified isatin molecules (1-(2-methylallyl)indoline-2,3-dione and 1-isobutylindoline-2,3-dione) and combining them with PABA, substituted o-aminobenzoic acids and p-aminomethylbenzoic acid, two series of Schiff base derivatives (23, 24) were obtained and evaluated for antibacterial, antifungal and antibiofilm potential [113]. The hybrids inhibited Gram-positive bacteria. 2-Methylallyl derivatives (23) presented greater activity than isobutyl counterparts (24), with the best action being recorded for 23a and 23b (S. aureus, B. subtilis—MIC 0.09 mmol/L, 0.181 mmol/L) compared with imipenem (MIC 0.036 mmol/L). This difference may be attributed to the fact that 2-methylallyl is less hydrophobic than isobutyl and is able to penetrate membranes more easily and form π-H interactions with protein targets. Compounds 23a ((E)-4-(1-(2-methylallyl)-2-oxoindolin-3-ylideneamino)benzoic acid) and 23b ((E)-2-(1-(2-methylallyl)-2-oxoindolin-3-ylideneamino)-4-chlorobenzoic acid) showed over 55% biofilm inhibition against S. aureus and MRSA compared to 20% for chloramphenicol. The docking study against B. subtilis histidine kinase/Walk YycG, the enzyme involved in biofilm formation and bacterial virulence [146], revealed the importance of hydrogen bonds between Asp105 and 23a for antibiofilm activity [113].
Schiff bases of methyl 12-aminooctadec-9-enoate (25a–f) were obtained and tested for antimicrobial and antibiofilm effects [114]. Gram-positive strains (S. aureus, B. subtilis) were susceptible to the action of the compounds, but Gram-negative bacteria remained resistant. The best antimicrobial effect was obtained for p-chloro derivative 25a, followed by hydroxy and methoxy azomethines (25c, 25d). All were inferior to ciprofloxacin (MIC 2.7 μM). N,N-dimethylamino and N,N-dimethyl cinnamyl Schiff bases presented moderate antibacterial activity (25b: MIC 17.6–35.2 μM, MBC 17.6–70.5 μM; 25e: MIC 16.6–33.3 μM, MBC 16.6–66.6 μM). The mechanism seems bactericidal due to MBCs closed to MICs (MBC/MIC < 4) [147]. Regarding antibiofilm action, the order was maintained as 25a, 25c, 25d and 25f having IC50 under 10 μM (ciprofloxacin IC50 0.99–1.53 μM) [114].
N-[(E)-4-bromo-2,5-diheptyloxybenzylideneamino]-2,4-dinitroaniline (26) was evaluated for antimicrobial, antibiofilm and antiquorum sensing activities at MIC and lower concentrations. E. faecalis and Candida strains were the most susceptible in terms of antimicrobial effect, while Gram-positive biofilms, especially of SA, were the most sensitive. It exhibited quorum sensing inhibition against C. violaceum and was able to reduce swarming motility of P. aeruginosa by 14.4–45.7% at MIC/4-MIC. Thus, it targets two steps in biofilm formation: communication and dispersion [115].
N-(2-hydroxybenzylidene)-2-hydroxypropanehydrazides (27a–f) and 2-hydroxy-N-((3-hydroxy-5-(hydroxymethyl)-2-methylpyridin-4-yl)-2-hydroxypropane-hydrazide (27g) were obtained by condensing (S)-lactic acid hydrazide with substituted salicylaldehydes or pyridoxal and tested for antibacterial and antibiofilm activities. The nitro derivative 27e was active against S. aureus, while the pyridoxal derivative 27g was able to inhibit E. coli (MIC 64 μg/mL for both derivatives and both strains). Also, they significantly reduced P. aeruginosa O1 biofilm formation at 1/16 and ¼ MIC [116].
2-Pyridinylhydrazone of substituted salicylaldehydes and pyridinylcarbaldehydes (28a–f) were active against A. baumannii planktonic and sessile cells. Electron-donating groups (R1: OH, OCH3) and nitro groups on salicylaldehydes moiety and 3 or 4-pyridinyl radical have a beneficial effect on antimicrobial and antibiofilm activities. All compounds were able to inhibit the biofilm of A. baumannii culture and clinical isolates (MIC < 200 μg/mL). Compounds 28a ((E)-3-((2-(pyridin-2-yl)hydrazono)methyl)benzene-1,2-diol) and 28d ((E)-4-nitro-2-((2-(pyridin-2-yl)hydrazono)methyl)phenol) exhibited the best antimicrobial activity (MIC 25 μg/mL) and acted as both biofilm inhibitors and disruptors (MIC < 25 μg/mL) [117].
Phenyl-2-(2-(1-phenylethylidene)hydrazinyl)thiazoles (29) were synthesized and evaluated against clinical isolates of C. albicans [118]. The thiazole ring was obtained in the reaction of thiosemicarbazones with substituted phenyl bromides. Biological assay revealed that compounds 29d–e and 29a–c were able to inhibit the biofilm formation at 50–100 μg/mL. Their action varied according to the substituents of the two benzene rings (R1, R2). Difluoro derivatives (29d–e) were equally potent, followed by methyl (29c), methoxy (29b) and unsubstituted (29a) analogues. Microscope imaging confirmed fungal biofilm formation reduction. Gene expression analysis indicated upregulation of inhibitory genes implicated in yeast-hyphae transition (bcy1, nrg1, tup1) and downregulation of genes responsible for C. albicans biofilm formation and virulence (als3, hwp1, ras1). The docking study indicated interactions between compounds and lanosterol 14-alpha-demethylase (van der Waals and hydrophobic bonds) [118].
Starting from sulfathiazole, antistaphylococcal compounds were obtained by the isostere replacement of the nitrogen atom of a sulphonamide fragment by methylene carbonyl group, resulting in 2-(4-aminobenzene-1-sulfonyl)-1-(1,3-thiazol-5-yl)ethan-1-ones [148]. Through the continued optimization of these structures, the carbonyl fragment was changed to the imine group, resulting in oximes, hydrazones and N-acylhydrazones analogues [119]. The imine group was introduced in order to improve solubility and antimicrobial potential, while radical R and R1 modulated activity. Biological assay revealed that the ethyl imine group is beneficial for activity. Acetylated derivatives are preferred over simple amines. N-acyl-hydrazones were the most active against tested strains, with hydrazones being favourable and oximes decreasing the activity (Figure 8). Compound 31f (N-(4-((2-(2-picolinoylhydrazono)-2-(thiazol-2-yl)ethyl)sulfonyl)phenyl)acetamide) was bactericidal against E. faecalis, standard and clinical isolates at MIC 1–4 μg/mL, surpassing norfloxacin (MIC 4–8 μg/mL) and sulfathiazole (MIC 128 μg/mL). It was also able to reduce the biofilm mass of E. faecalis by 35% at 6xMIC. Some possible mechanisms of action for compound 31f are membrane damage, oxidative damage, inhibition of dihydrofolate synthetase and complexation of DNA [119].
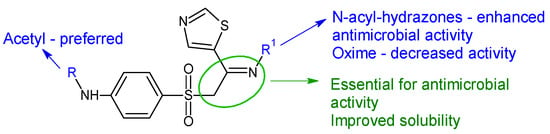
Figure 8. Structure–activity relationship for compounds 30, 31.
(E)-N’-((5-nitrofuran-2-yl)methylene)quinoline-8-sulfonohydrazide (32) was obtained combining the pharmacophores 5-nitro-furan and quinoline and linked via a sulfonyl–hydrazone bond. Antimicrobial screening revealed antifungal potential against culture type fungal strains and clinical isolates (MIC 125–250 μg/mL) and modest antibacterial properties. The compound was able to inhibit C. albicans at 32.1 μg/mL, yeast-hyphae transition at 24.96 μg/mL and fungal biofilm formation (38% inhibition at MIC). Quinoline and furan rings form hydrophobic and aromatic interactions with the active site of Als3, while the nitro group interact with Tyr21 via hydrogen bonds [120].
Three N’-(1-(3-hydroxynaphthalen-2-yl)ethylidene)sulfonohydrazides (33a–c) were evaluated for antifungal and antibiofilm action against collection strains and clinical isolates. They surpassed fluconazole (MIC 128 μg/mL) in the case of clinical isolates of C. albicans and C. krusei (MIC 32 μg/mL). For the same strains, they inhibited biofilm formation at 32–64 μg/mL. All compounds downregulate hyphae-specific genes hwp1, als3 and ece1, and 33b and 33c also reduced the expression of sap5 genes, with propyl derivative being the most potent [121].
Schiff bases (34a–e) derivatives of 2-((hydrazinocarbonyl)methoxy)-4-phenyl-6-(2-thienyl)pyridine-3-carbonitril were synthesized and evaluated for antimicrobial and antibiofilm activity [122]. Compounds 34a (2-((benzylidene-hydrazinocarbonyl)methyloxy)-4-phenyl-6-(2-thienyl)pyridine-3-carbonitrile) and 34c (2-((4-methoxy-benzylidene-hydrazinocarbonyl)methyloxy)-4-phenyl-6-(2-thienyl)pyridine-3-carbonitrile) were moderately active against E. coli planktonic and biofilm forms (IR 64.81, 64.61%, BI 78.75, 73.67%, respectively). The isatin derivative 34e presented moderate antistaphylococcal activity and antibiofilm activity against P. aeruginosa (inhibition ratios over 60%). The chloro derivative 34d had reduced or no effect [122].
A series of six (EZ)-N’-benzylidene-(2RS)-2-(6-chloro-9H-carbazol-2yl)-propanhydrazides (35a–f) were synthesized and tested for antibacterial, antifungal and antibiofilm activities [123]. Gram-positive bacteria (S. aureus, E. faecalis), as well as C. albicans, were sensitive to the action of the compounds, with MICs reaching 0.15–0.31 mg/mL for 35a, 35c and 35d. The antibiofilm activity was similar, with compound 35c inhibiting C. albicans biofilm at 0.009 mg/mL while compound 35d acted on S. aureus, E. faecalis (MBIC: 0.078 mg/mL). 4-chloro substitution was beneficial for antibacterial and antifungal activity and hydroxy enhanced antistaphylococcol action, whereas the 3,5-dichloro derivative 35f was inactive [123].
Hydrazones of 5-hydroxy-2,2-dimethyl-2H-chromene-6-carbaldehyde with different aryl, sulfonyl and non-aryl hydrazines were obtained and evaluated for QS inhibition and antibacterial activity [124]. Sulfonyl derivative ((E)-N’-((5-hydroxy-2,2-dimethyl-2H-chromen-6-yl)methylene)benzenesulfonohydrazide—36f) and semicarbazone (36a) exhibited moderate anti-QS activity (IC50 22 μM, respectively 27 μM), but no antibacterial effect against V. harveyi. Substitution on the sulfonyl ring with hydrophobic groups (methyl, trifluoromethyl) or changing the urea to thiourea abolished anti-QS effect (Figure 9). Compounds 36d (4-OH), 36c (H), 36e (2,4-diOH), 36i and 36j (pyridyl) presented antibacterial activity against V. harveyi (MIC 3.9, 7.8, 10.0, 10.0, 15.6 μM). Compound 36e was the only one active against S. aureus (MIC 64 μg/mL) without effect on E. coli [124].
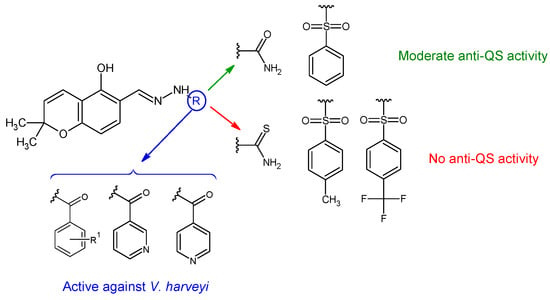
Figure 9. Structure–activity relationship for compounds 36.
Starting from 4-[4-formyl-3-(2-naphthyl)pyrazol-1-yl]benzoic acid [126] and 4-[3-(7-fluoro-2-oxo-3,8a-dihydrochromen-3-yl)-4-formyl-pyrazol-1-yl]benzoic acid [125], two series of hydrazones have been obtained. The selected derivatives (37a–d, 38a–e) exhibited antimicrobial activity against Gram-positive bacteria comparable to vancomycin (MIC 0.195-3.125 μg/mL). 37b and 37c inhibited A. baumannii as well. All compounds had the ability to inhibit S. aureus biofilm. 37a,c,d reached over 85% inhibition and 38c over 90% inhibition at 1/2 MIC, which is better than vancomycin (>60% inhibition at 1/2 MIC). They also disrupted the preformed biofilm—37a–c over 90% and 38b,e over 70% at 1/2MIC.
Schiff base derivatives of androstane-1,4-diene-3,17-dione, thiosemicarbazone (39a) and isonicotinoylhydrazone (39b) presented antifungal and fungal biofilm inhibition properties. Both compounds surpassed ketoconazole (MIC 0.20–1.00 mg/mL) in some instances in terms of antifungal action, with thiosemicarbazone 39a being the most potent. Compound 39b also presented a higher binding affinity towards CYP51 of C. albicans than ketoconazole, interacting with Fe of heme. However, they were inferior biofilm inhibitors (ketoconazole: BI 25–55%), with compound 39a performing slightly better than 39b [127].
5-Nitro-2-thiophenecarbaldehyde N-[(E)-(5-nitrothienyl)methylidene)hydrazone (40) was evaluated for antistaphylococcal activity [128]. It inhibited Pan-S S. aureus at 0.5–2.0 μg/mL, VRSA and MRSA. Exposing the biofilm to this compound for 24 h led to a noteworthy (p < 0.05) decrease in the integrity of S. aureus biofilm at a concentration 4× MIC. The findings indicate that this hydrazone can impact S. aureus biofilm integrity even at concentrations 10–40× MIC. Additional research is necessary to gain a deeper understanding of the mechanism behind the disruption of S. aureus biofilm and potential interactions with biofilm-targeting properties of 40 and other antimicrobials available in clinical settings.
(E)-1H-indole-3-carbaldehyde O-(4-chlorobenzyl)oxime, (E)-1H-indole-3-carbaldehyde O-(4-bromobenzyl) oxime (41a,b) and (E)-1-(1H-indol-3-yl)ethan-1-one O-(3,4-dichlorobenzyl)oxime (41c) presented antistaphylococcal activity (1–8 μg/mL) against standard and drug resistant strains (VRSA, MRSA). Biofilm inhibition capacity was reduced (10% at 1–10× MIC) comparable to references (levofloxacin and vancomycin) [129].
Antistaphylococcal furanoquinone derivatives (oximes, hydrazones) (42, 43) were synthesized starting from naphto[2,3-b]furan-4,9-dione and naphto[1,2-b]furan-4,5-dione [130]. Based on biological evaluation, structure–activity relationships revealed that naphto[1,2-b]furan-4,5-dione is essential for activity, while linear furanoquinones are inactive. Oxime group (X: O) is necessary for MRSA inhibition, with small radicals (R: H- 43a, COCH3- 43b) being favoured over bulky substituents. Phenyl radicals coupled with hydrazine linker (X: N) showed moderate activity and were inferior to oximes (Figure 10). Thus, (Z)-4-(hydroxyimino)naphtho[1,2-b]furan-5(4H)-one (43a) and (Z)-4-(acetoxyimino)naphtho[1,2-b]furan-5(4H)-one (43b) were the most active of the series, being active against planktonic and sessile forms of MRSA. They exhibited bactericidal action against S. aureus standard strains, drug resistant strains and clinical isolates. They were able to penetrate MRSA biofilm and completely inhibit bacteria outside the matrix at 100 μg/mL, surpassing cetylpyridinium chloride. Bacteria inside the matrix were less susceptible, with a reduction of 4-log CFU being observed for hydroxy at 100 μg/mL. Biofilm height was reduced to half by both compounds. They also presented activity against MRSA-infected wounds with minimum skin irritation. The mechanism of action seemed to be inhibition of DNA gyrase (43a, 43b) and RNA polymerase (43a) [130].
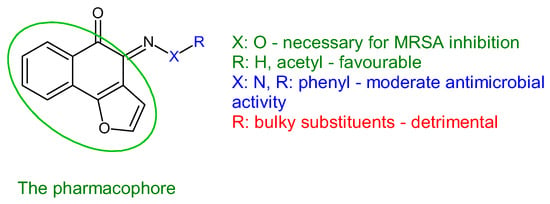
Figure 10. Structure–activity relationship for compounds 43.
2-Methyl-l1-hydroxyimino-6,11-dihydrodibenzo[b,e]thiepin-5,5-dioxide (44a) and 2-methyl-l1-hydroxyimino-6,11-dihydrodibenzo[b,e]thiepin-5,5-dioxide (44b) [131] demonstrated microbicidal activity against the Gram-negative, non-fermentative A. baumanii. These oximes effectively hindered the adherence ability of C. albicans strains to inert substrata at a concentration of 250 µg/mL. Additionally, they displayed notable antibiofilm activity against the Gram-negative, non-fermentative bacilli P. aeruginosa and A. baumanii. Molecular modelling suggests that these compounds may interfere with the synthesis of quorum sensing molecules, specifically N-acyl-l-homoserine lactones, utilized by Gram-negative strains as their potential targets. It’s worth noting that despite the absence of fungicidal activity, compounds 44a and 44b exhibited inhibitory effects on the development of fungal biofilms.
Tetrahydroberberine, a natural alkaloid, was combined with metronidazole, a narrow-spectrum antimicrobial, and with oxime fragments to yield a series of derivatives (45a–j). These derivatives were subsequently tested for their antimicrobial and antibiofilm activities [55]. The hybrids demonstrated enhanced potency and a broader spectrum in comparison to berberine and metronidazole. The antimicrobial activity was influenced by the radical R of the oxime component. Linear alkyl groups (methyl to hexyl, 45b–h) had a detrimental effect, whereas branched tert-butyl (45f), unsaturated allyl (45i) and benzyl (45j) were found to be beneficial. Simple oxime 45a (9-(2-hydroxy-3-(2-methyl-5-nitro-1H-imidazol-1-yl)propoxy)-10-methoxy-5,8,13,13a-tetrahydro-6H-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinoline-12-carbaldehyde oxime) was active against Gram-positive bacteria (S. aureus, E. faecalis), Gram-negative bacteria (E. coli, P. aeruginosa, A. baumannii) and fungi C. albicans, C. parapsilosis, A. fumigatus (MIC 0.029–0.058 mM). Compound 45j (9-(2-hydroxy-3-(2-methyl-5-nitro-1H-imidazol-1-yl)propoxy)-10-methoxy-5,8,13,13a-tetrahydro- 6H-[1,3]dioxolo[4,5-g]isoquinolino[3,2-a]isoquinoline-12-carbaldehyde O-benzyl oxime) had remarkably low MIC values (0.024–0.199 mM), especially on Gram-negative strains, P. aeruginosa in particular (0.024 mM), surpassing in some instances norfloxacin. It was also able to reduce P. aeruginosa biofilm in a dose-dependent manner (45% inhibition at 8× MICs) and seemed to act against bacterial cell membrane. Regarding antifungal assay, 45j inhibited all fungal strains except C. parapsilosis (MIC 0.024–0.199 mM) [132].
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics13010075
This entry is offline, you can click here to edit this entry!