Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
The hospital mortality in patients with ST-segment elevation myocardial infarction (STEMI) is about 6% and has not decreased in recent years. The leading cause of death of these patients is ischemia/reperfusion (I/R) cardiac injury. It is quite obvious that there is an urgent need to create new drugs for the treatment of STEMI based on knowledge about the pathogenesis of I/R cardiac injury, in particular, based on knowledge about the molecular mechanism of ferroptosis.
- ferroptosis
- heart
- ischemia/reperfusion
- kinases
- cardiomyopathy
- microRNAs
1. The Main Manifestation of Ferroptosis
Ferroptosis ends with cell membrane rupture and intracellular protein release [10,11,12,13]. However, intracellular protein release occurs in necrosis, necroptosis, and pyroptosis [14,15,16]. Lipid peroxidation is accompanied by malondialdehyde (MDA), reactive oxygen species (ROS), and 4-hydroxynonenal (4-HNE) formation [10,11,17]. In one study, the MDA level was increased by 30–300% [17,18,19,20,21,22]. The 4-HNE level was increased by 40% [18]. Glutathione (GSH) was reduced by 70–97% in ferroptosis [18,19,23]. Some protein expression has also shown to be altered in ferroptosis. In other studies, the expression of acyl-CoA synthetase long-chain family member 4 (ACSL4) was increased by 130–300% [19,21,23]. Elsewhere, the expression of prostaglandin endoperoxide synthase 2 (PTGS2) was increased by 200–300% [21,24]. In some studies, the expression of the transferrin receptor (TFR-1) was reduced by 30% [18] or increased 2-fold [19,25]. The expression of cystine/glutamate transporter (SLC7A11) (xCT) was reduced by 90% [18]. In other studies, ferritin heavy chain-1 (FTH1) expression was decreased by 70–170% [19,24]. Elsewhere, the glutathione peroxidase-4 (GPX4) level was decreased by 40–70% [19,20,22,23,24,25]. However, some investigators could not find alterations in the GPX4 level in ferroptosis [18]. None of the listed markers are specific for ferroptosis. Therefore, investigators usually evaluate the four or five marker levels [17,19,21,23,24,25,26]. An important indicator of the involvement of ferroptosis in a pathological process is a decrease in its intensity after the use of deferoxamine, ferrostatin-1, UAMC-3203, dexrazoxane, and liproxstatin-1 which are ferroptosis inhibitors [12,27,28].
2. Inhibitors of Ferroptosis
The first articles in which the Fe2+ chelator deferoxamine was considered an inhibitor of ferroptosis were published 10 years ago [29,30]. However, the first articles which considered deferoxamine as an inhibitor of lipid peroxidation were published over 50 years ago [31,32]. In 2012, the first article that demonstrated the ability of ferrostatin-1 to inhibit ferroptosis was published [12]. In 2014, the first article that demonstrated the ability of liproxstatin-1 to inhibit ferroptosis was published [33]. The ability of UAMC-3203 to inhibit ferroptosis was later demonstrated by Devisscher et al. [34]. Ferrostatin-1 stability in rat plasma (% recovery after 6 h) is 1.1% [34]. UAMC-3203 stability in rat plasma (% recovery after 6 h) is 100% [34]. Therefore, UAMC-3203 is more effective in a long-term study than ferrostatin-1. In 2009, it was reported that the Fe2+ chelator dexrazoxane can mitigate anthracycline cardiotoxicity [35]. The investigators suggested that dexrazoxane prevents cardiomyocyte death, which is triggered by “ROS and iron”, after the application of anthracycline. Later, dexrazoxane was considered a pharmacological tool for studying ferroptosis [36]. It was reported that dexrazoxane can cross cell membranes and reduce the intracellular free Fe2+ level [28]. At present, deferoxamine, ferrostatin-1, UAMC-3203, liproxstatin-1, and dexrazoxane are used in the study of ferroptosis.
3. The Role of Kinases in the Regulation of Ferroptosis
It has been demonstrated that the activation of AMP-activated protein kinase (AMPK), extracellular signal-regulated kinase 1/2 (ERK1/2), phosphoinositide 3-kinases (PI3K), Akt kinase, protein kinase C (PKC), NO synthase (NOS), heme oxygenase-1 (HO-1), cyclooxygenase-2 (COX-2), and Janus kinase-2 (JAK2) promote an increase in cardiac tolerance to ischemia/reperfusion (I/R) [37]. In contrast, the stimulation of c-Jun N-terminal kinases (JNKs) and glycogen synthase kinase-3β (GSK-3β) contributes to a decrease in cardiac tolerance to I/R [37]. It could be hypothesized that these enzymes regulate the ferroptosis of cardiomyocytes.
In one study, mice were subjected to CAO (60 min) and reperfusion (24 h) [38]. According to Gomez et al. (2008), an intravenous administration of the GSK-3β inhibitor SB216763 prior to reperfusion contributed to a decrease in infarct size by about 34% [38]. Ischemic postconditioning exhibited the same infarct-reducing effect [38]. The investigators argue that the infarct-reducing effect of postconditioning is a consequence of the phosphorylation (inactivation) of GSK-3β [38].
AMPK. It was found, in another study, that ferulic acid reduced infarct size in rat with coronary artery occlusion (CAO, 30 min) and reperfusion (120 min) [39]. Ferulic acid simultaneously inhibited ferroptosis in myocardial tissue [39]. Pretreatment with compound C, an AMPK inhibitor, abolished the inhibition of ferroptosis and the infarct size reduction in rats [39]. Glycation end-products stimulated ferroptosis in isolated rat cardiomyocytes [40]. Ferrostatin-1 and deferoxamine inhibited ferroptosis. This effect was eliminated by compound C [40]. Consequently, AMPK is involved in the cytoprotective effect of ferroptosis inhibitors. One study reported that Puerarin, an active ingredient in the traditional Chinese medicine Pueraria, inhibited lipopolysaccharide (LPS) and induced myocardial ferroptosis in rats [19]. This effect was abolished by compound C [19]. Consequently, AMPK is involved in inhibition of ferroptosis. Another study found that ischemia/reperfusion induced ferroptosis in an isolated rat heart [41]. The α2-adrenergic receptor (α2-AR) agonist, dexmedetomidine, suppressed ferroptosis. This effect was abolished by compound C [41]. According to one study, canagliflozin, a sodium–glucose cotransporter-2 inhibitor, alleviated palmitic acid-induced ferroptosis of the HL-1 cardiomyocyte cell line [42]. Compound C eliminated this effect of canagliflozin. Another study reported that embryonic rat heart-derived H9c2 cells were exposed to H2O2 which induced ferroptosis of these cells [43]. Idebenone, an analog of coenzyme Q10 (CoQ10), mitigated ferroptosis. The investigators obtained evidence that AMPK could be involved in the inhibition of ferroptosis [43]. In another study, it was found that CAO (30 min) and reperfusion (24 h) induced ferroptosis in a rat heart [44]. Britanin, a bioactive sesquiterpene lactone isolated from Inula lineariifolia, reduced infarct size, alleviated ferroptosis, and increased the p-AMPK level in myocardial tissue [44]. These data demonstrated the involvement of AMPK in inhibition of ferroptosis (Figure 1).
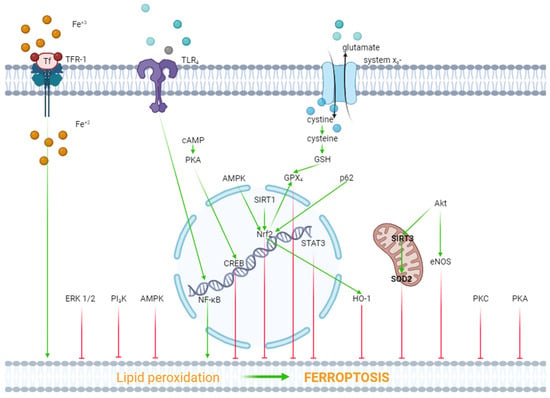
Figure 1. The role of kinases and transcription factors in the regulation of ferroptosis. Red arrows are inhibitors of ferroptosis, and green arrows are inducers of ferroptosis.
ERK1/2. In one study, it was reported that hypoxia/reoxygenation (H/R) induced ferroptosis of H9c2 cells [45]. Dexmedetomidine alleviated ferroptosis, increased cell viability, and triggered the phosphorylation (activation) of ERK1/2. The inhibition of ERK1/2 by U0126 reversed the cytoprotective effect of dexmedetomidine and mitigated the dexmedetomidine-triggered suppression of ferroptosis [45]. Consequently, ERK1/2 is involved in the inhibition of ferroptosis (Figure 1).
Protein kinase A (PKA). As it have already reported above, H/R induced ferroptosis of H9c2 cells [45]. Dexmedetomidine partially reversed this effect. The PKA inhibitor H89 eliminated the inhibition of ferroptosis by dexmedetomidine. SiRNA against CREB also partially reversed the dexmedetomidine-triggered inhibition of ferroptosis, where CREB is a cAMP response element-binding protein. The investigators concluded that dexmedetomidine alleviated H/R injury of H9c2 cells by suppressing ferroptosis through the activation of the cAMP/PKA/CREB signaling pathway (Figure 1).
PKC. One study found that doxorubicin and erastin, a ferroptosis inducer, resulted in ferroptosis of H9c2 cells [46]. Pretreatment with the E-prostanoid 1 receptor agonist 17-PT-PGE2 increased cell viability and inhibited ferroptosis [46]. The PKA and PKC inhibitor staurosporine (20 nM/L) reversed the 17-PT-PGE2-triggered inhibition of ferroptosis [46]. It should be noted that staurosporine, at a final concentration of 20 nM/L, completely blocks PKC and partially inhibits PKA [47,48]. Consequently, it could be argued that the activation of PKC promotes the inhibition of ferroptosis (Figure 1).
Akt. In one study, H9c2 cells were subjected to H/R [49]. H9c2 cells were transfected with a microRNA miR-199a-5p inhibitor to down-regulate miR-199a-5p and an miR-199a-5p mimic to up-regulate miR-199a-5p prior to H/R. The miR-199a-5p inhibitor increased cell viability, suppressed ferroptosis, and increased the p-Akt/Akt ratio [49]. The Akt inhibitor LY294002 abolished the cytoprotective effect of the miR-199a-5p inhibitor and reduced the p-Akt/Akt ratio [49]. Consequently, the activation of Akt promotes the inhibition of ferroptosis and an increase in cell viability in H/R. In another study, it was found that doxorubicin induced a cardiotoxic effect which is accompanied by ferroptosis [22]. LCZ696, an angiotensin receptor and neprilysin inhibitor, protects the rat heart against doxorubicin and suppresses ferroptosis. The Akt inhibitor LY294002 alleviates the cardioprotective and anti-ferroptotic effects of LCZ696. Investigators have suggested that sirtuin-3, a soluble mitochondrial NAD-dependent deacetylase, is involved in the cardioprotective effect of LCZ696. It has been shown that LCZ696 protects H9c2 cells against the cytotoxic effect of doxorubicin and inhibits doxorubicin-induced ferroptosis. Sirtuin-3 knockout abolishes both protective effects of LCZ696. In addition, these investigators found that LCZ696 stimulates the expression of superoxide dismutase-2 (SOD2). They concluded that the cardioprotective effect of LCZ696 is mediated via the activation of the Akt/sirtuin-3/SOD2 pathway [22]. Thus, the stimulation of Akt alleviates ferroptosis of cardiomyocytes (Figure 1).
NOS. The researchers have already reported above that the miR-199a-5p inhibitor suppressed ferroptosis and increased H9c2 cell survival in H/R through the activation of Akt [49]. It was found that the miR-199a-5p inhibitor increased the concentration of NO in a culture supernatant of H9c2 cells [49]. The miR-199a-5p inhibitor increased the p-eNOS/eNOS ratio. The Akt inhibitor LY294002 abolished an increase in the p-eNOS level. The investigators concluded that the miR-199a-5p inhibitor increased H9c2 cell tolerance to H/R through the stimulation of the Akt/eNOS pathway. However, the anti-ferroptotic effect of canagliflozin is accompanied by a decrease in the inducible NOS (iNOS) mRNA level in HL-1 cells [42]. Thus, there is no definition of the role of NOS in the regulation of ferroptosis in myocardial tissue (Figure 1).
PI3K. One study reported that doxorubicin induced the death and ferroptosis of H9c2 cells [23]. Pretreatment with 740Y-P, a PI3K activator, mitigated both effects of doxorubicin and increased HO-1 expression [23]. Lapatinib, an ErbB-2 and EGFR tyrosine kinase inhibitor, enhanced doxorubicin-induced ferroptosis and reduced the p-Akt level in H9c2 cells [23]. In another study, it was found that trastuzumab, an anticancer drug, induced ferroptosis of cardiomyocytes both in vivo and in vitro and also reduced the p-PI3K/PI3K ratio [50]. Ferrostatin-1 and deferoxamine inhibited ferroptosis of cardiomyocytes and increased the p-PI3K/PI3K ratio [50]. It has also been reported that suberosin exhibits cardioprotective and anti-ferroptotic effects which are associated with an increase in the PI3K mRNA in rats pretreated with the ferroptosis inducer thiazolidinedione [51]. Suberosin is a natural product that is isolated from the roots and aerial parts of Cudrania tricuspidata [51]. These data demonstrate that the stimulation of PI3K promotes the inhibition of ferroptosis (Figure 1).
COX-2. Zhang et al. (2023) did not find convincing evidence of the involvement of COX-2 in the regulation of palmitic acid-induced ferroptosis in HL-1 cells [42].
HO-1. Doxorubicin-induced ferroptosis is associated with an increase in the HO-1 mRNA level in murine hearts [36]. Sepsis-induced ferroptosis is accompanied by an increase in HO-1 expression in the murine heart [52]. The cardioprotective effect of the α2-AR agonist dexmedetomidine in mice with sepsis has been associated with a decrease in HO-1 expression in the murine myocardium [52]. One study reported that sickle cell disease induced cardiomyopathy and ferroptosis in the murine heart and promoted the upregulation of HO-1 in myocardial tissue [53]. The inhibition of HO-1 by tin protoporphyrin-IX caused the suppression of ferroptosis in mice with SCD. In contrast, the induction of ferroptosis promoted HO-1 expression in mice [53]. In another study, it was found that the chronic administration of di(2-ethylhexyl) phthalate (DEHP) induced ferroptosis in the murine heart [54]. This effect is associated with an increase in HO-1 expression in myocardial tissue. One study reported that doxorubicin induced ferroptosis and increased HO-1 expression in HL-1 cells [55]. HMOX1 knockdown vector (HMOX1 short hairpin RNA (shRNA)) reduced HO-1 expression and inhibited the ferroptosis of HL-1 cells [55]. Cardiac-specific Sirtuin 1 knockout aggravated the cardiotoxic effect of doxorubicin and ferroptosis in mice [55]. Both effects were accompanied by an increase in HO-1 expression in myocardial tissue [55]. It was reported elsewhere that the HO-1 inhibitor zinc protoporphyrin suppressed isoproterenol-induced myocardial ferroptosis [56]. It was found, in another study, that the cytoprotective and anti-ferroptotic effects of the MiR-432-5p mimic are associated with an increase in the HO-1 level in isolated cardiomyocytes subjected to H/R [57].
These data demonstrate that HO-1 is involved in the pathogenesis of ferroptosis of cardiomyocytes (Figure 1).
GSK-3β. It was shown, in one study, that britanin reduced infarct size, alleviated ferroptosis, and increased the p-GSK-3β level in myocardial tissue of rats with I/R of the heart [58]. Bian et al. (2023) showed that palmitic acid induced ferroptosis of human cardiomyocyte AC16 cells [59]. This effect was associated with a reduction in the phosphorylation of Akt and GSK-3β. Celastrol, a bioactive compound isolated from the herb Tripterygium wilfordii, inhibited ferroptosis and increased cell viability. Celastrol simultaneously triggered the phosphorylation of Akt and GSK-3β. The investigators suggested the Akt/GSK-3β signaling pathway participated in the anti-ferroptotic and cytoprotective effects of celastrol [59]. According to Gomez et al. (2008), the phosphorylation-induced inactivation of GSK-3β plays a negative role in cardiac tolerance to reperfusion [38]. Consequently, a decrease in the phosphorylation (activation) of GSK-3β could promote ferroptosis. Bian et al. (2023) [59] did not provide an explanation for this discrepancy and did not discuss Gomez’s data. Consequently, the role of GSK-3β in the regulation of ferroptosis requires further study.
In summary, these data demonstrate that the activation of AMPK, HO-1, ERK1/2, PKA, PKC, Akt, and PI3K promotes the inhibition of ferroptosis. In contrast, the simulation of GSK-3β contributes to the ferroptosis of cardiomyocytes.
4. The Role of Non-Coding RNA in the Regulation of Ferroptosis in the Heart
In recent years, much attention has been paid to studying the role of non-coding RNAs (ncRNAs), particularly microRNAs, long non-coding RNAs (lncRNAs), and circular RNAs in the pathogenesis of cardiovascular diseases, in particular, in the regulation of ferroptosis [60,61].
It was found, in one study, that lncRNA Snhg7 plasmid induced ferroptosis of -1 cells via the activation of T-box transcription factor 5 (Tbx5) [62]. It was demonstrated, in another study, that the serum level of small extracellular vesicle-encapsulated (SEMA5A-IT1) RNAs negatively correlated with the serum creatine kinase-MB (CK-MB) level in patients with a cardiopulmonary bypass [63]. SEMA5A-IT1 RNAs are lncRNAs. Human cardiomyocyte AC16 cells were exposed to H/R. The cells were transfected by the lentiviral vectors of SEMA5A-IT1. These lncRNAs increased cell survival, inhibited apoptosis and ferroptosis through an increase in miR-143-3p expression. The miR-143-3p mimic exhibited the same cytoprotective effects as lncRNAs [63].
In one study, erastin, a ferroptosis inducer, induced ferroptosis and the death of H9c2 cells [64]. H9c2 cells were transfected with a lentiviral vector expressing miR-190a-5p which inhibits GLS2 gene expression (this gene encodes the synthesis of glutaminase 2). miR-190a-5p overexpression increased cell viability and inhibited ferroptosis. In contrast, anti-miR-190a-5p decreased cell survival and enhanced erastin-induced ferroptosis [64]. In a different study, mice underwent permanent CAO for 3 days [65]. CAO induced an increase in the miR-15a-5p level 2-fold. The investigators suggested that miR-15a-5p could regulate the tolerance of cardiomyocytes to H/R [65]. HL-1 cells were exposed to hypoxia for 24 h which induced the death of 30% of cells. MiR-15a-5p aggravated hypoxia-induced cell death through a reduction in GPX4 expression and an increase in the MDA and ROS levels in HL-1 cells. The investigators concluded that miR-15a-5p could be involved in the development of I/R cardiac injury through the activation of ferroptosis [65]. One study reported that Erastin induced ferroptosis and the death of HL-1 cells [66]. It was found that circRNA1615 reduced the cytotoxic effect of erastin. Investigators have proposed that the cytoprotective effect of circRNA1615 is a result of its anti-ferroptotic effect [64]. In a different study, rats underwent permanent CAO [67]. The duration of CAO was 28 days. An adverse remodeling of the heart was developed which was accompanied by the activation of ferroptosis. CAO induced an increase in miR-375-3p content in myocardial tissue by approximately 4-fold. It was found that miR-375-3p inhibited GPX4 expression. Ferrostatin-1 and the miR-375-3p inhibitor suppressed ferroptosis and improved the contractility of the heart. The investigators suggested that miR-375-3p induced ferroptosis through the inhibition of GPX4 expression, and the miR-375-3p inhibitor alleviated this process and prevented the adverse remodeling of the heart [67]. It have been already reported above that the miR-199a-5p inhibitor increased H9c2 cell viability and suppressed ferroptosis in H/R through the activation of Akt [49]. In one study, cultured rat cardiac microvascular endothelial cells (CMEC) were subjected to hypoxia [19]. Exosomes were isolated from the incubation medium of CMEC and added to H9c2 cells exposed to H/R. H/R induced ferroptosis and the death of H9c2 cells. The exosomes increased cell viability and inhibited ferroptosis. These exosomes contained miR-210-3p. The exosomes inhibited erastin-induced cell death and ferroptosis. The miR-210-3p inhibitor abolished the cytoprotective and anti-ferroptotic effects of exosomes. In another study, the miR-210-3p mimics suppressed ferroptosis [21]. Elsewhere, cultured human cardiac myocytes were subjected to hypoxia (1% O2) for 24 h [68]. Hypoxic cardiomyocytes secreted exosomes containing miR-208a/b. Erastin induced ferroptosis of cultured human cardiac fibroblasts (CFs). Exosomes enhanced erastin-induced ferroptosis of CFs. miR-208a/b inhibitors reversed the pro-ferroptotic effect of exosomes. The investigators concluded that hypoxic cardiomyocyte-derived exosomes can aggravate ferroptosis of CFs through miR-208a/b expression [68]. In a different study, exosomes isolated from the plasma of mice with permanent CAO inhibited erastin-induced ferroptosis and increased the survival of the Lewis lung carcinoma cell line LLC and osteosarcoma cell line K7M2 [69]. These exosomes contained miR-22-3p. This microRNA inhibited erastin-induced ferroptosis and increased tumor cell viability [69]. These data demonstrate that miR-22-3p is an inhibitor of ferroptosis.
One study showed that hypoxia induced ferroptosis and the death of H9c2 cells [44]. It was found that miR-26b-5p mimics aggravated hypoxia-induced cell death and stimulated ferroptosis of H9c2 cells [44]. In a different study, the miR-214-3p level was increased in the infarcted region of the murine heart and in neonatal rat cardiomyocytes (NRCMs) subjected to hypoxia [70]. An increase in miR-214-3p content is accompanied by ferroptosis, and in this study, the miR-214-3p inhibitor (antagomir) improved cardiac contractility, reduced infarct size, and alleviated ferroptosis in myocardial tissues. Consequently, miR-214-3p induced ferroptosis of NRCMs. The miR-214-3p inhibitor protected NRCMs against hypoxia. The investigators suggested that malic enzyme 2 is a target of miR-214-3p. They proposed that miR-214-3p is an endogenous trigger of ferroptosis which suppresses malic enzyme-2 expression [70].
In one study, lipopolysaccharide from Escherichia coli induced sepsis-like cardiomyopathy in mice [71]. miR-130b-3p overexpression improved the contractility of the septic heart, reduced the serum creatine kinase-MB (CK-MB) and cardiac troponin I (cTnI) levels, and inhibited ferroptosis in myocardial tissue. LPS induced ferroptosis and the death of H9c2 cells. The miR-130b-3p mimic inhibited ferroptosis and increased cell viability [71]. In contrast, the miR-130b-3p inhibitor decreased cell viability and stimulated ferroptosis [71]. H/R caused an increase in the circ_0091761 RNA level in H9c2 cells. As found in one study, ferrostatin-1 resulted in a decrease in lactate dehydrogenase (LDH) release, decreased circ_0091761 expression, and inhibited ferroptosis of H9c2 cells [72]. The circ_0091761 inhibitor (si-circ_0091761) increased H9c2 cell viability and suppressed ferroptosis. H/R caused an increase in miR-335-3p content in H9c2 cells [72]. Si-circ_0091761 increased miR-335-3p expression in H9c2 cells in H/R. miR-335-3p mimics increased cell viability and inhibited ferroptosis. The investigators concluded that circ_0091761 enhanced H/R-induced cell death and ferroptosis and that circ_0091761 and an miR-335-3p mimic could protect the heart against I/R [72]. Elsewhere, HL-1 cells were exposed to hypoxia (1% O2) for 18 h [73]. Hypoxia reduced miR-450b-5p content in HL-1 cells and induced ferroptosis. miR-450b-5p mimics increased cell viability, reduced cTnI release from HL-1 cells, and suppressed ferroptosis in these cells [73].
In one study, neonatal rat ventricular cardiomyocytes were exposed to H/R [57]. H/R induced cell death and ferroptosis. An miR-432-5p mimic plasmid increased cell viability and inhibited ferroptosis [57]. The cytoprotective effect of the miR-432-5p mimic was associated with an increase in the expression of nuclear factor erythroid 2-related factor 2 (Nrf2). In addition, the miR-432-5p mimic increased HO-1 expression in cardiomyocytes and decreased it in the Kelch-like ECH-associated protein 1 (Keap1) protein level. It was reported that Keap1 is an endogenous inhibitor of Nrf2 [57]. It was found that miR-432-5p-Lipo reduced infarct size by about 30% and inhibited ferroptosis in myocardial tissue in rats with CAO (30 min) and reperfusion (4 h) [57]. The investigators concluded that the miR-432-5p mimic inhibits ferroptosis through the activation of Nrf2 and HO-1 expression in cardiomyocytes and the inhibition of Keap1 expression [57].
Thus, circ_0091761 RNA, lncRNA Snhg7, miR-214-3p, miR-199a-5p, miR-208a/b, miR-375-3p, miR-26b-5p and miR-15a-5p can aggravate ferroptosis. In contrast, miR-190a-5p, circRNA1615, miR-22-3p, miR-450b-5p, miR-130b-3p, miR-335-3p, miR-432-5p, miR-143-3p, SEMA5A-IT1 RNAs and miR-210-3p can inhibit ferroptosis. These data demonstrate that miR-450b-5p, miR-432-5p and miR-210-3p can increase the tolerance of cardiomyocytes to hypoxia/reoxygenation through the inhibition of ferroptosis. Circ_0091761 RNA, miR-214-3p, miR-199a-5p, miR-375-3p, miR-26b-5p, miR-335-3p, and miR-15a-5p can aggravate H/R-induced injury of cardiomyocytes through the enhancement of ferroptosis (Figure 2).
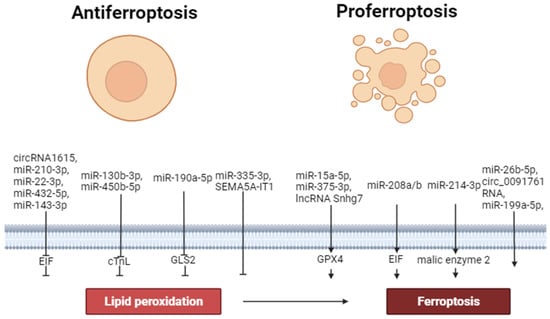
Figure 2. The role of non-coding RNAs in the regulation of ferroptosis. Perpendicular arrows are inhibitors of ferroptosis, arrows are inducers of ferroptosis.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25020897
This entry is offline, you can click here to edit this entry!