Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Respiratory System
Extracellular lysophospholipids (lysophosphatidic acid, lysophosphatidylcholine, sphingosine 1-phosphate, etc.), which are synthesized from phospholipids in the cell membrane, act as lipid mediators, and mediate various cellular responses in constituent cells in the respiratory system, such as contraction, proliferation, migration, and cytoskeletal organization. In addition to these effects, the expression of the adhesion molecules is enhanced by these extracellular lysophospholipids in pulmonary endothelial cells. These effects are exerted via specific G protein-coupled receptors.
- mRNA
- fatty acid
- ATX
1. Structure and Function
Lysophosphatidic acid is the simplest bioactive phospholipid that is synthesized in the cell membranes of various tissues including in endothelial cell, and it exerts potent extracellular signaling through its interaction with its six specific GPCRs as a lipid mediator, resulting in cell proliferation, migration, contraction, and cytoskeletal reorganization. LPA consists of a glycerol backbone with the addition of a phosphate group at the sn-3 position and a hydroxyl group and a fatty acid chain in either the sn-1 or sn-2 position, with variations being due to different fatty acid chains. The most common LPA form known in research is described as 18:1 (with an 18-carbon chain length and 1 unsaturated bond). LPA receptors (LPA6) recognize ligands (LPA) through conformational rearrangement (inward shift of transmembrane helices 6 and 7) [28]. Extracellular LPA is produced from lysophospholipids, such as LPC and LPS, by the enzymatic activity of extracellular autotoxin (ATX), which has lysophospholipase D activity and is classified as ectonucleotide pyrophosphatase-phosphodiesterase 2 (ENPP2), and it is also produced from PA by PA-selective PLA1. ATX is derived from the epithelium and macrophages. In addition to this process, LPA is generated from PA by PLA1/PLA2 independent of ATX. ATX is generally considered to be a major source of generated LPA, and extracellular LPA plays an essential role in physiological activities as a lipid mediator via cellular responses through LPA receptors. LPA receptors are widely expressed in several organs, including the brain and lungs. The activation of LPA1,2, which is connected to Rho/Rho-kinase processes, causes contraction in muscle cells via an increase in the response to intracellular Ca2+ (Ca2+-independent contraction), referred to as Ca2+ sensitization [29,30]. Since Rho/Rho-kinase processes also cause the dynamic reorganization of the actin cytoskeleton in non-muscle cells, the activation of LPA1,2 is associated with alterations in cell motility and morphology, resulting in cell migration and the dysfunction of cell–cell adhesion. Therefore, LPA causes phenotype changes in constituent cells of the respiratory system, i.e., smooth muscle contractility, fibroblast and inflammatory cell recruitment, epithelial apoptosis, and endothelial permeability [24]. A recent report has indicated that LPA5 may be a novel target for the development of agents for inflammatory and malignant diseases [31].
2. Effects on Smooth Muscle Cells
Since the Rho/Rho-kinase pathway is the intracellular signal transduction system of LPA receptors, except for LPA3, smooth muscle in the respiratory system may generate tension via Rho-mediated Ca2+ sensitization in the presence of LPA (Ca2+-independent contraction). A previous report has demonstrated that LPA enhances the contractility of isolated tracheal smooth muscle in rabbits and cats [32]. However, little is currently known about the contractile effects of LPA on pulmonary smooth muscle in detail. LPA generates tension in endothelium-denuded aorta segments [33]; moreover, LPA prevents endotoxin-induced dilatations in the pulmonary artery via the disassembly of the smooth muscle cell (SMC) F-actin cytoskeleton via the activation of the Rho/Rho-kinase pathway [34] (Figure 1), suggesting that LPA may cause contraction in pulmonary vascular smooth muscle, similar to airway smooth muscle [32]. Since the PLC/IP3 pathway is the intracellular signal transduction system of LPA receptors, except for LPA6, LPA may cause the contraction of pulmonary vascular smooth muscle via Ca2+ dynamics (Ca2+-dependent contraction). LPA increases the intracellular Ca2+ concentration in cultured A10 vascular smooth muscle cells through store-operated Ca2+ entry (SOCE) via the PLC/IP3 pathway, a Na+-Ca2+ exchanger, and a Na+-H+ exchanger [11] (Figure 1). However, it is debatable whether LPA’s directly induced contraction in pulmonary vascular smooth muscle is involved in the pathophysiology of pulmonary hypertension. LPA enhances proliferation and migration in the vascular smooth muscle cells of the aorta and carotid artery [35,36,37] (Figure 1). Blood vessel restenosis is associated with proliferation and migration in vascular smooth muscle cells, which may be mediated by LPA during restenosis. When proliferation and migration are investigated using an electrical cell–substrate impedance sensor (ECIS), an inhibitor of ATX (PF8380) and an inhibitor of LPA receptors (Ki16425) attenuate proliferation and migration in carotid arterial smooth muscle cells in mice [37]. Cell proliferation and cell migration in smooth muscle cells may cause pulmonary arterial remodeling, resulting in pulmonary hypertension.
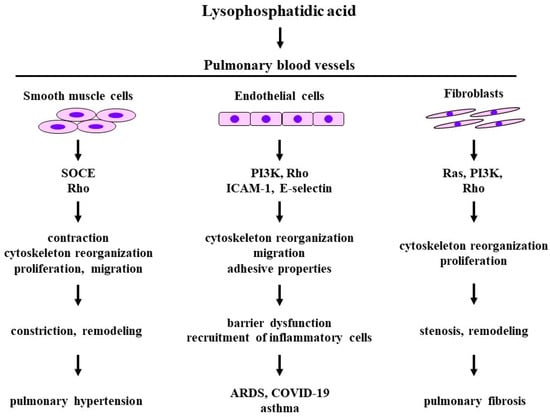
Figure 1. The effects of lysophosphatidic acid on the constituent cells and related molecular mechanisms in pulmonary blood vessels. Disease arising from pathophysiology based on physiological activities induced by lysophosphatidic acid. SOCE: store-operated calcium entry; PI3K: phosphoinositol 3-kinase; ICAM-1: intercellular adhesion molecule 1. The 1st row: the constituent cells; the 2nd row: molecular mechanisms; the 3rd row: biological activities; the 4th row: pathophysiology; the 5th row: diseases related to pulmonary vasculature. Illustrated based on Refs. [5,9,11,14,15,17,24,29,30,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48].
3. Effects on Fibroblasts
LPA causes cytoskeletal reorganization and the proliferation of lung fibroblasts through GPCRs-mediated signal pathways, such as Rho, Ras, and PI3K, resulting in pulmonary fibrosis and pulmonary hypertension [24] (Figure 3). Both the serum levels of LPA and the perivascular expression of LPA are increased in a rat model of hypoxic pulmonary hypertension, which is characterized by structural changes in the vascular wall of pulmonary arteries [14]. Moreover, the expression of ATX is increased in coexistence with mast cell tryptase in these hypoxic rat lungs, indicating that mast cells are involved in the generation of LPA. Augmented ATX staining is observed in the lungs of bleomycin-treated mice, and ATX levels are also elevated in corresponding BALFs [38]. Serum from low-oxygen-challenged rats has higher chemoattractant effects on primary lung fibroblasts, indicating that LPA facilitates fibroblast migration [14]. Since this phenomenon is suppressed in the presence of inhibitors of LPA1,3, LPA contributes to hypoxia-induced remodeling in the pulmonary vasculature, resulting in pulmonary hypertension [14]. In LPA1-lacking mice, the development of pulmonary fibrosis is markedly prevented, and the absence of LPA1 causes a decrease in fibroblast recruitment, indicating that LPA1 causes pulmonary fibrosis via fibroblast recruitment [15]. This phenomenon may be excessive when injury leads to fibrosis rather than to repair.
4. Effects on Endothelial Cells
An aberrant vascular system is a characteristic feature of ATX knockout mice, indicating that LPA may be involved in the malformation of vasculature during development [39]. LPA is associated with the function of endothelial cells via the expression of angiogenesis and the modulation of their permeability [40]. These endothelial cell functions are impaired by LPA-induced barrier dysfunction, leading to cancer and various inflammatory diseases [41] (Figure 3). The LPA-induced barrier function in the endothelium is caused by Rho-mediated cytoskeletal reorganization due to stress fibers which consist of actin filaments. LPA1 expressed in endothelial cells is probably associated with increased vascular permeability in lung injury [15] (Figure 1). Endothelial cell migration, which is an essential component of angiogenesis and wound repair, is activated by LPA. Since this phenomenon is inhibited by pretreatment with pertussis toxin (an agent for the uncoupling of Gi from receptors) and LY294002 (an inhibitor of PI3K), endothelial cell migration induced by LPA is associated with the Gi/PI3K pathway, which causes actin filament remodeling (cytoskeletal reorganization) through Akt [42,43] (Figure 1).
LPA also markedly augments the adhesive properties of endothelial cells in the pulmonary artery and mononuclear cells in the peripheral blood in a concentration-dependent manner. The effects of LPA on adhesive properties are markedly attenuated in the presence of VPC-12249, an antagonist of LPA1,2 [14]. The pre-exposure of human pulmonary arterial endothelial cells to LPA also enhances the mRNA expression of adhesion molecules such as intracellular adhesion molecule 1 (ICAM-1), E-selectin, β1 integrin, and vascular cell adhesion molecule 1 (VCAM-1) [14] (Figure 1). LPA-induced expression of adhesion molecules in the endothelium may result in the recruitment of inflammatory cells in the respiratory system. Inhalation of LPA into guinea pigs augments the number of neutrophils and eosinophils in BALFs in a concentration-dependent manner, and inhalation of LPA enhances superoxide synthesis by these inflammatory cells compared with spontaneous production [17]. These effects of LPA on inflammatory cell recruitment and superoxide synthesis are attenuated in the presence of Y-27632, an inhibitor of Rho-kinase, in a concentration-dependent manner, indicating the involvement of the Rho/Rho-kinase pathway in these phenomena.
5. Clinical Relevance
5.1. Pulmonary Hypertension
LPA is associated with various inflammatory diseases based on vascular dysfunction via the physiological action on endothelial and smooth muscle cells [44]. It is still unknown whether pulmonary smooth muscle cells are involved in LPA-induced pulmonary vascular remodeling. However, in a rat model of hypoxic pulmonary hypertension, LPA levels are markedly increased in serum and tissues, and fibroblast migration and the recruitment of mononuclear cells are facilitated in vessels in the respiratory system, resulting in pulmonary vascular remodeling [14] (Figure 1). Pulmonary vasculature remodeling probably causes hypertension in the pulmonary artery.
5.2. Pulmonary Fibrosis
A clinical trial has demonstrated that LPA levels are markedly increased in patients with idiopathic pulmonary fibrosis, and that the suppression of LPA1 markedly attenuates the chemotactic effects on the fibroblasts in this fluid [15] (Figure 1). LPA is increased in BALFs and exhaled breath condensates in patients with idiopathic pulmonary fibrosis (IPF), and the expression of ATX with lysophospholipase D activity that converts LPC into LPA is increased in lung tissues from patients with IPF and fibrotic non-specific interstitial pneumonia [38] (Figure 1). The up-regulation of ATX is also closely associated with more progressive and irreversible forms of pulmonary fibrosis, such as IPF/usual interstitial pneumonia (UIP) and fibrosing nonspecific interstitial pneumonia (fNSIP) [38]. ATX-LPA is probably involved in the pathogenesis of pulmonary fibrosis. Moreover, the knockdown of LPA2 causes a reduction in bleomycin-induced lung injury and pulmonary fibrosis, and this phenomenon may be associated with a reduction in the LPA-induced expression of TGF-β and the activation and differentiation of fibroblasts [45]. Therefore, LPA1,2 is probably a novel therapeutic target for idiopathic pulmonary fibrosis, which responds to injury implicated in fibrosis.
5.3. Asthma
When patients with asthma are challenged with an allergen, ATX/LPA levels are augmented in their BALFs, indicating that ATX/LTA may be involved in the allergic reaction related to asthma [9]. The inhibition of ATX causes a marked reduction in Th2 cytokines and allergic inflammation in the respiratory system in murine model of allergic asthma [46]. The antigen-challenged BALFs of ATX transgenic mice have a markedly increased number of inflammatory cells (mostly eosinophils) and Th2 cytokine levels (IL-4, IL-5) compared with wild-type mice; in contrast, mice in which the ATX-LPA pathway in blocked have a markedly decreased number of eosinophils and Th2 cytokine levels compared to wild-type mice [46]. Therefore, the ATX-LPA pathway may be a novel therapeutic target for asthma. The ATX-LPA pathway enhances superoxide levels [37], suggesting that LPA may be involved in the pathophysiology of asthma and chronic obstructive pulmonary disease (COPD), which are partly related to oxidative stress [47].
5.4. Acute Respiratory Distress Syndrome: COVID-19
ATX levels in serum are increased in patients with severe COVID-19; increased serum levels of ATX are corelated with increased serum levels of IL-6, as well as endothelial dysfunction markers, such as soluble E-selectin, soluble P-selection, ICAM-1, and angiopoietin 2 [5] (Figure 1). Severe COVID-19 could lead to the development of ARDS with impaired gas exchange. ATX levels in serum and BALFs are associated with inflammatory and fibrotic biomarkers (IL-6, IL-8, TNF-a, fibronectin, etc.), and the severity of illness (the SOFA score and PaO2/FIO2 ratio) in patients with ARDS, indicating that ATX/LPA may be involved in the pathogenesis of ARDS [48]. The ATX-LPA pathway could be involved in the cytokine storm and subsequent pulmonary fibrosis in the pathophysiology of COVID-19. LPA-induced barrier dysfunction in endothelial cells may be involved in the pulmonary edema related to ARDS.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines12010124
This entry is offline, you can click here to edit this entry!