Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Cofilin is an essential protein in cytoskeletal dynamics, and any dysregulation could lead to potentially serious complications. Cofilin’s involvement is underscored by its impact on pathological hallmarks like Aβ plaques and α-synuclein aggregates, triggering synaptic dysfunction, dendritic spine loss, and impaired neuronal plasticity, leading to cognitive decline.
- cofilin
- neuroinflammation
- neurodegenerative diseases
- cofilin inhibition
- cofilin signaling
1. Introduction
Neurodegenerative diseases represent an elusive and growing global health challenge, imposing an ever-increasing burden on individuals, families, and healthcare systems [1]. These diseases, characterized by the progressive degeneration and loss of neurons, manifest in a multitude of forms, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and stroke [2]. Despite their distinct etiologies, these conditions share common underlying cellular and molecular mechanisms contributing to neuronal dysfunction and demise [3]. In this era of intensive neurodegeneration research, cofilin protein has emerged as one of the essential players in the intricate web of neuronal homeostasis and dysfunction [4]. Cofilin, a highly conserved actin-binding protein, has garnered significant attention for its pivotal role in modulating cytoskeletal dynamics, cell motility, and synaptic plasticity [5,6]. Beyond its fundamental cellular functions, mounting evidence has revealed that cofilin has been implicated in neurodegenerative disease pathogenesis, opening up new possibilities for understanding and potentially intervening in the disease [7,8,9].
2. Cofilin: Structure and Function
2.1. Molecular Structure and Regulation of Cofilin
Cofilin, a vital actin-binding protein, is ubiquitously expressed in eukaryotic cells and plays a fundamental role in regulating cytoskeletal dynamics [13]. It comes under a group of proteins collectively known as the actin-depolymerizing factor (ADF)/cofilin family, encompassing ADF, also recognized as destrin, along with cofilin-1, the predominant isoform found ubiquitously in non-muscle tissues, and cofilin-2, the primary isoform in mature muscle tissues [14,15] (Figure 1). For this review, our focus will be on cofilin-1, referred to henceforth as cofilin. Its molecular structure and intricate regulatory mechanisms are vital to understanding its multifaceted functions within the cell [16]. Cofilin is a small protein consisting of approximately 166 amino acids. It is highly conserved across species, underscoring its essential role in cellular processes. Cofilin’s secondary structure consists of alpha helices and beta strands. These elements fold and arrange themselves to form specific structural motifs. The secondary structure contributes to the protein’s overall stability and three-dimensional architecture [17]. At the tertiary level, cofilin adopts a well-defined structure. It comprises a central core region enriched in alpha-helical structures surrounded by beta strands. This compact and organized tertiary structure enables cofilin to interact with actin filaments with precision [18].
The primary structure of cofilin contains key functional domains, including the N-terminal actin-binding domain and the C-terminal regulatory domain [19,20]. The N-terminal domain of cofilin is responsible for binding to filamentous actin (F-actin) [21]. Cofilin binds preferentially to ADP-bound actin monomers, leading to conformational changes in F-actin and promoting filament severing and depolymerization [22].
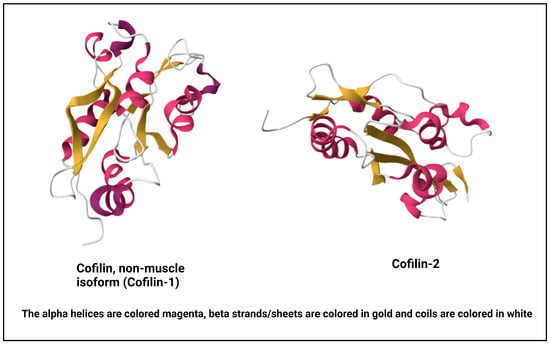
Figure 1. NMR structure of human cofilin isoforms [23,24,25]. (https://doi.org/10.2210/pdb1Q8G/pdb; https://doi.org/10.2210/pdb7M0G/pdb, (accessed on 2 January 2024)).
This activity is central to regulating actin dynamics within the cell [26]. The C-terminal region of cofilin contains phosphorylation sites critical for its regulation. The activity of cofilin is tightly regulated through a balance of phosphorylation and dephosphorylation events [27]. These regulatory mechanisms are crucial in modulating cofilin’s effects on the actin cytoskeleton. Serine-3 (Ser-3) is the primary phosphorylation site, and its phosphorylation status dictates cofilin’s activity [28]. LIM kinases (LIMKs) phosphorylate cofilin at Ser-3, leading to its inactivation [27,28,29]. This phosphorylation prevents cofilin from binding to F-actin, thereby inhibiting its ability to promote actin filament depolymerization [30]. Slingshot phosphatases, such as SSH1 and SSH2, dephosphorylate cofilin at Ser-3, restoring its activity. This dephosphorylation allows cofilin to bind to actin filaments, sever them, and enhance actin dynamics [31]. Apart from phosphorylation/dephosphorylation, cofilin’s activity is also pH-sensitive. It exhibits increased actin-binding and severing activity at lower pH levels, particularly relevant in cellular compartments with varying pH, such as synaptic terminals [32,33,34]. Moreover, other actin-binding proteins, such as profilin and tropomyosin, can modulate cofilin’s interaction with actin filaments, further fine-tuning its effects on cytoskeletal dynamics [35,36,37,38]. Understanding cofilin’s molecular structure and regulation is pivotal in deciphering its diverse cellular functions, including its involvement in neuronal processes, cell migration, and synaptic plasticity [39]. Dysregulation of these regulatory mechanisms can have profound implications for cellular function and has been linked to pathological conditions, including neurodegenerative diseases [40,41,42].
2.2. Role of Cofilin in Different Cellular Processes
Cofilin, a crucial regulator of actin cytoskeletal dynamics, plays a multifaceted role in various physiological processes, encompassing cell cycle regulation, cell motility, cell structure maintenance, autophagy, and apoptosis [13]. Cofilin helps in cytokinesis during cell division by contributing to contractile ring formation and dissolution, ensuring proper separation of daughter cells [43]. At the forefront of cell migration, cofilin actively promotes lamellipodia formation, facilitating the assembly and extension of thin, sheet-like protrusions rich in actin filaments. By severing and depolymerizing actin filaments, it contributes to dynamic lamellipodia remodeling, enabling cell movement and migration efficiency [44]). During cell migration, cofilin helps in lamellipodia formation and contributes to focal adhesions, which anchor the cell to the extracellular matrix and must undergo dynamic turnover. Cofilin contributes to this process by modulating actin dynamics at focal adhesion sites. It helps disassemble focal adhesions at the cell’s rear, allowing cells to detach and move forward [27]. Cofilin’s role in the motility of growth cones is particularly crucial in the context of neuronal systems, as growth cones are specialized structures at the tips of developing neurites (axon and dendrite growth processes) that play a fundamental role in guiding and navigating axons during neural development [45]. Cofilin’s involvement in growth cone motility is tightly linked to its ability to regulate the dynamic rearrangements of the actin cytoskeleton, a process essential for neurite outgrowth and guidance. The growth cone’s motility relies on the dynamic assembly and disassembly of actin filaments [46]. Actin filaments at the leading edge of the growth cone undergo polymerization, pushing the growth cone forward. At the same time, actin filaments at the rear of the growth cone undergo depolymerization, allowing forward movement [47]. By enhancing actin filament turnover, cofilin increases the pool of available actin monomers for filament assembly, promoting dynamic actin remodeling within the growth cone. Furthermore, cofilin’s activity is spatially and temporally regulated within the growth cone. At the leading edge, where active neurite outgrowth occurs, cofilin is involved in promoting actin dynamics. In contrast, cofilin may contribute to actin depolymerization at the central and rear regions, facilitating neurite retraction or turning. Growth cones navigate through chemotactic responses, where guidance cues direct their movement [48]. Cofilin-mediated actin dynamics are integral to the growth cone’s ability to interpret and respond to these guidance cues, allowing for precise axon pathfinding. Understanding the detailed molecular mechanisms of cofilin in growth cone motility provides insights into the fundamental processes that underlie neuronal development [49]. Dysregulation of cofilin activity in this context can have profound implications for axon guidance, neurite outgrowth, and the establishment of neural circuits, potentially contributing to neurodevelopmental disorders or compromised neuronal regeneration in the adult nervous system [50]. Cofilin also participates in intracellular trafficking by influencing the movement of vesicles, organelles, and other cargo within the cell, thus regulating the actin cytoskeleton’s organization [9]. This role of cofilin extends to the dynamic regulation of AMPA receptors. AMPA receptors are crucial for synaptic transmission and plasticity, and their proper trafficking is essential for maintaining synaptic function [51]. AMPA receptors mediate the majority of fast excitatory neurotransmissions in the central nervous system. Proper trafficking of AMPA receptors to and from the synapse is essential for synaptic plasticity, a process crucial for learning and memory [52]. Actin dynamics, regulated by proteins like cofilin, play a pivotal role in orchestrating these trafficking events. Dysregulation of actin-dependent AMPA receptor trafficking can contribute to excitotoxicity, a phenomenon where excessive activation of glutamate receptors leads to neuronal damage and death. This process is implicated in several pathological conditions, including AD and stroke [8,41]. In AD, aberrant AMPA receptor trafficking and excitotoxicity have been associated with synaptic dysfunction and neurodegeneration. Similarly, in stroke, the disruption of normal cellular processes, including AMPA receptor trafficking, contributes to excitotoxic neuronal damage following ischemic events [53].
Moreover, cofilin is vital for immune cell function, including chemotaxis, phagocytosis, and immune synapse formation. It enables immune cells to respond effectively to pathogens and foreign substances [54]. For an efficient chemotaxis effect, cofilin helps promote the flow of cytoplasmic material toward the cell’s leading edge, facilitating directed cell movement [55]. Particularly in immune cell migration and metastatic cancer cell invasion, cofilin’s actin cytoskeleton remodeling ability allows the cells to squeeze through tight spaces, a process known as amoeboid or bleb-based motility [30]. In neurons, it is involved in synaptic plasticity (a process essential for learning and memory) by impacting the remodeling of dendritic spines and, consequently, synaptic strength and morphology [39,56,57]. Dysregulation of cofilin can affect various cellular processes and has been implicated in pathological conditions, making it a significant focus of research in the context of neurodegenerative diseases and stroke. The following sections focus on the link between cofilin dysregulation and the pathophysiology of various neurodegenerative diseases and stroke.
3. Cofilin in Neurodegeneration
As previously mentioned, initially recognized for its pivotal role in cytoskeletal dynamics and cell motility, cofilin has emerged as a critical factor in developing neurodegenerative diseases [9]. Multiple lines of evidence connect cofilin dysregulation to these conditions, highlighting its contributions to neuronal dysfunction and degeneration. In AD, two major pathological features, amyloid-β (Aβ) and hyperphosphorylated tau, have been implicated in cofilin dysregulation. Aβ promotes cofilin dephosphorylation and activation, leading to excessive actin depolymerization, dendritic spine loss, synaptic dysfunction, and cognitive decline [5,41]. Hyperphosphorylated tau can also activate cofilin, contributing to the formation of neurofibrillary tangles and disruption of neuronal cytoskeletal structure [58]. In PD, α-synuclein, a key player in the disease’s pathogenesis, interacts with cofilin, potentially influencing actin dynamics and synaptic dysfunction [59]. Additionally, mitochondrial dysfunction in PD activates cofilin, leading to mitochondrial fragmentation and neuronal damage [60]. HD, characterized by the accumulation of mutant huntingtin protein, interacts with and activates cofilin. This interaction results in abnormal actin dynamics, neuronal cytoskeletal abnormalities, and synaptic dysfunction in HD-affected neurons [61]. Another neurodegenerative disease, amyotrophic lateral sclerosis (ALS), associated with TDP-43 protein pathology, affects cofilin phosphorylation and activity, contributing to cytoskeletal abnormalities and motor neuron degeneration [62].
In ischemic stroke, widespread neuronal injury and neuroinflammation is observed. Cofilin activation is implicated in ischemia-induced neuronal death and dendritic spine loss, adding to the neurological damage [9,63]. In the case of traumatic brain injury (TBI), axonal injury is a hallmark feature. Cofilin activation leads to axonal damage by promoting actin filament severing and destabilization [64,65]. Cofilin dysregulation has also been explored in various other neurodegenerative conditions, including frontotemporal dementia, multiple sclerosis, and prion diseases, suggesting its potential as a common pathological mechanism [4,66,67,68,69].
The evidence linking cofilin to neurodegenerative diseases underscores its role in mediating cytoskeletal dysfunction, synaptic impairment, and neuronal degeneration. Dysregulation of cofilin activity, often driven by alterations in its phosphorylation status, disrupts the delicate balance of actin dynamics in neurons, leading to dendritic spine loss, axonal damage, and impaired synaptic plasticity. This disruption plays a pivotal role in the pathophysiology of neurodegenerative diseases. Understanding the molecular mechanisms underlying cofilin dysregulation in these conditions is an active area of research, holding promise for developing targeted therapies aimed at preserving neuronal integrity and function in the face of neurodegeneration.
4. Cofilin Dysregulation in Stroke and Other Neurodegenerative Disorders
Ischemic stroke is a devastating neurological condition characterized by the sudden interruption of blood flow to the brain, resulting in the oxygen and nutrient deprivation of neurons [70]. Cofilin plays a significant role in the pathogenesis of stroke and other neurodegenerative diseases [71]. Here, we provide an overview of cofilin’s involvement in stroke and other neurodegenerative diseases and its implications for neuronal damage and neuroinflammation (Figure 2).
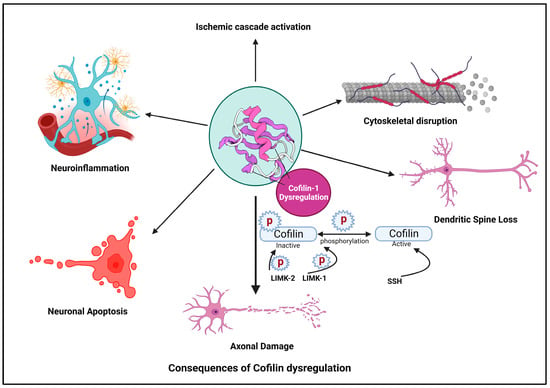
Figure 2. Cofilin dysregulation leads to neuroinflammation and neuronal apoptosis.
4.1. Ischemic Cascade
In response to ischemia, various cellular pathways are activated, leading to neuronal injury and death. Cofilin is a critical participant in these cascades [72]. Hypoxia and energy depletion following ischemia can lead to the dephosphorylation and activation of cofilin, promoting actin depolymerization [73].
4.2. Cytoskeletal Disruption
Activated cofilin can disrupt the actin cytoskeleton in neurons [40]. The activation of cofilin induces cytoskeletal disruption primarily through its ability to modulate actin dynamics. When cofilin is activated, it binds to actin filaments, which are the structural components of the cytoskeleton. Cofilin exhibits a preference for ADP-actin subunits within these filaments. Cofilin induces a conformational change in actin filaments, promoting the severing of these filaments into smaller fragments [74]. This process increases the number of free barbed ends essential for actin polymerization. Activated cofilin enhances the depolymerization of actin filaments by increasing the rate at which actin monomers dissociate from the filament ends [75]. This results in a higher turnover of actin filaments within the cytoskeleton. The increased depolymerization leads to a surplus of free globular (G)-actin monomers within the cellular pool, making them available for new filament assembly [76]. The combined actions of cofilin result in a shift in the dynamic equilibrium of actin filaments. There is an overall increase in the turnover of actin filaments, leading to a more dynamic and less stable actin cytoskeleton [77].
Loss of Cytoskeletal Integrity: The continuous severing and depolymerization induced by activated cofilin disrupt the structural integrity of the actin cytoskeleton. This dynamic instability can affect various cellular structures, including dendritic spines, synapses, and overall neuronal morphology. Cytoskeletal disruption contributes to neuronal damage and impairs neuronal functions [78].
4.3. Dendritic Spine Loss
Dysregulated cofilin activity, marked by its activation and subsequent influence on actin dynamics, plays a pivotal role in disrupting dendritic spine morphology and function [39]. Under normal conditions, cofilin regulates actin filament turnover, maintaining the structural plasticity of dendritic spines. However, when activated, cofilin enhances actin severing and depolymerization, destabilizing the actin cytoskeleton within spines [5]. This disruption extends to critical actin-dependent processes, including endocytosis and receptor recycling. Cofilin-induced alterations in actin dynamics impact the formation of endocytic vesicles, influencing the internalization of membrane components and synaptic receptors [79]. Concurrently, the compromised actin structure leads to abnormal dendritic spine morphology, characterized by filopodial protrusions and enlarged heads. Weakened actin filaments render dendritic spines susceptible to structural collapse, resulting in spine loss. This synaptic disconnection, attributed to dysregulated cofilin activity, contributes to impaired synaptic transmission and holds implications for neurodegenerative conditions, such as AD [5,41,80]. Ischemia-induced cofilin activation has been linked to dendritic spine loss in affected neurons [8]. This spine loss compromises synaptic connectivity and contributes to cognitive deficits observed in stroke patients [81].
4.4. Axonal Damage
Cofilin activation can promote the severing of actin filaments in axons, leading to axonal fragmentation and damage [82]. Axonal pathology, characterized by axonal swellings and spheroids formation, is a common feature in conditions like TBI and ALS [83]. Axonal damage disrupts neuronal communication and can lead to functional deficits [84]. Cofilin activation can lead to axonal damage in response to ischemia [8]. The severing of actin filaments in axons contributes to axonal fragmentation and impaired neuronal communication [85]. Axonal damage may further exacerbate neurological deficits and impair functional recovery post-stroke [72].
4.5. Neuronal Apoptosis
Dysregulated cofilin activity can trigger apoptotic pathways in neurons [86]. The cytoskeletal instability induced by cofilin activation can damage DNA and activate pro-apoptotic proteins [87]. Neuronal apoptosis contributes to losing viable neurons in the ischemic penumbra and core regions of the stroke-affected brain [88].
4.6. Neuroinflammation in Stroke, Its Implications and Cofilin as a Mediator of Neuroinflammatory Responses
Neuroinflammation plays a dual role in stroke, contributing to both protective and detrimental effects [89]. Chronic neuroinflammation is a hallmark of several neurodegenerative diseases and can exacerbate neuronal damage by activating immune cells and inducing oxidative stress [90,91,92]. Ischemic stroke triggers an immune response in the brain, activating immune cells such as microglia and attracting infiltrating leukocytes, including neutrophils and monocytes. Cofilin plays a crucial role in activating immune cells in the brain, influencing their migration, phagocytosis, and antigen presentation. This activation is prompted by the release of damage-associated molecular patterns (DAMPs) from injured neurons and astrocytes [93,94]. These activated immune cells release pro-inflammatory mediators, including cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-1β, chemokines, and reactive oxygen species (ROS) [95,96]. These inflammatory signals contribute to the recruitment of more immune cells and exacerbate neuronal damage [97,98]. It also impacts actin dynamics in immune cells, contributing to processes such as immune synapse formation and phagocytosis [99]. Cofilin activation in astrocytes can also contribute to neuroinflammation by promoting the release of pro-inflammatory cytokines and chemokines [90]. Additionally, cofilin indirectly influences blood–brain barrier (BBB) integrity, facilitating immune cell infiltration into the brain. Neuroinflammation can also disrupt the BBB, increasing its permeability. This allows immune cells to more easily infiltrate the brain and permits potentially harmful molecules, including pathogens and toxins, to enter [63,100]. Moreover, Cofilin’s activity is involved in the dynamic regulation of tight junctions between endothelial cells. Tight junctions are critical for forming a barrier that restricts the passage of substances between blood and brain tissue [101]. Cofilin’s ability to promote actin filament turnover influences the assembly and disassembly of tight junctions, thereby contributing to the permeability characteristics of the BBB. Cofilin-mediated actin dynamics are essential for the cytoskeletal remodeling of endothelial cells. This remodeling is necessary for processes such as cell migration, which is crucial during BBB development and repair [101,102]. Furthermore, neuroinflammation contributes to secondary brain injury, which extends beyond the initial ischemic insult and can persist for hours to days after the stroke. This secondary injury phase is characterized by ongoing neuronal death, edema, and an expansion of the infarct area [103,104,105]. Crucial roles are played by glial cells, including microglia and astrocytes, during neuroinflammation. Microglia transition to a pro-inflammatory state, producing cytokines and ROS, while reactive astrocytes release pro-inflammatory molecules, further propagating the inflammatory response [106,107,108]. In the neuroprotective role of inflammation during stroke, anti-inflammatory signals and immune cells are recruited to the injured area to dampen the inflammatory response, limiting further damage and promoting tissue repair and functional recovery [109,110,111]. The duration and intensity of neuroinflammation have significant implications for stroke recovery and rehabilitation outcomes. Excessive or prolonged inflammation may hinder recovery efforts, making it a challenging aspect of post-stroke care to balance the inflammatory response while promoting neuroprotection and neuroplasticity [12,112,113].
Studies have shown that the formation of cofilin-actin rods post-reperfusion underscores the dynamic response of neurons to ischemic insult, leading to disruptions in the cytoskeletal architecture and, consequently, impairments in organelle transport and dendritic spine loss. The increase in cofilin-1 levels, observed in the penumbra of the motor cortex following ischemia-reperfusion, implicates its potential role in gait imbalance and neuronal structural reorganization. The alterations in cofilin-1 mRNA and protein levels underline a complex regulatory mechanism during cerebral ischemia-reperfusion, potentially involving transcriptional and post-translational processes [96].
As previously mentioned, cofilin mediates neuroinflammatory responses in various neurological conditions, including stroke [8]. This involvement of cofilin in neuroinflammation highlights its potential as a therapeutic target for modulating neuroinflammatory processes. Understanding the role of cofilin in neuroinflammation is essential for exploring potential therapeutic strategies to modulate this complex process. Therefore, understanding the complexities of neuroinflammation has led to the development of various strategies, including immunomodulatory agents, anti-inflammatory drugs, and interventions to reduce BBB permeability. Emerging research explores anti-inflammatory approaches in combination with neuroprotective therapies [63,114,115,116]. A comprehensive understanding of the mechanisms involved in neuroinflammation is crucial for developing targeted therapies that harness its beneficial aspects while mitigating its detrimental effects, ultimately improving outcomes for stroke survivors.
This entry is adapted from the peer-reviewed paper 10.3390/cells13020188
This entry is offline, you can click here to edit this entry!