Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Heart failure (HF) with preserved ejection fraction (HFpEF) is an increasingly frequent form and is estimated to be the dominant form of HF. On the other hand, HFpEF is a syndrome with systemic involvement, and it is characterized by multiple cardiac and extracardiac pathophysiological alterations.
- heart failure
- HFpEF
- pathophysiological mechanism
- phenotypes
- treatment
1. Introduction
The diagnosis and treatment of heart failure (HF) with preserved ejection fraction (HFpEF) represent one of the greatest challenges for physicians today.
Although HFpEF has been seen as a mild condition in terms of organ damage, when it comes to treating these patients, there has been limited progress in developing an effective therapy. Perhaps this situation is due to the fact that HFpEF is a systemic syndrome with multi-organ involvement, which corroborates multiple cardiac and extracardiac physiopathological alterations [3,4,5].
HFpEF was defined by Dr. Luchi et al. in 1982, being the first group of researchers to describe typical heart failure symptoms in a group of patients with preserved left ventricular (LV) ejection fraction (EF) [6]. Recently, the European Society of Cardiology (ESC) united under the term HFpEF patients with preserved left ventricular EF (LVEF ≥ 50%) but with evidence of diastolic dysfunction or structural heart disease, classic signs and symptoms of heart failure and elevated plasma natriuretic peptide (NP) levels [1].
As with any definition of a syndrome, there are limitations, one of which is related to the lack of congestion in compensated HF. Another limitation is represented by the group of patients with HF symptoms who present abnormal hemodynamics exclusively during physical exercises [7]. HFpEF has been seen as a low-impact condition, yet patients have symptoms, signs, and quality of life not much different from those of patients with heart failure with reduced ejection fraction (HFrEF). Expert opinions support that HFpEF is rather a heterogeneous syndrome that includes different phenotypes with a spectrum of distinct, overlapping characteristics [8]. The etiology of HFpEF is unclear, and probably often multifactorial, but several culprits have been identified: microvascular lesions, low-grade systemic inflammation, and general oxidative stress (evolving in the context of comorbidities associated with endothelial dysfunction), all of these leading to myocardial remodeling and fibrosis [9]. These detrimental elements seem to participate fundamentally in the pathogenesis of the disease [8].
2. Pathophysiology of HFpEF
2.1. Left Ventricular Structure and Remodeling
The initial model indicated for HFpEF in descriptive studies was that of a ventricle of normal size but with hypertrophied walls (concentric left ventricular hypertrophy) [55]. Like any model, this one was not representative of all patients with HFpEF, some of them not having cardiac structural remodeling, the left ventricular geometry being normal. However, most patients with HFpEF correspond to the previously exposed pattern, recognized by the following characteristics: hypertrophy by increasing left ventricle (LV) wall thickness and/or LV mass and end-diastolic volume within normal or near-normal limits. Hypertrophy can be concentric hypertrophy by increasing the ratio of myocardial mass to cavity volume or generalized hypertrophy by increasing relative wall thickness (RWT) [56]. There is also a group of patients who present eccentric hypertrophy, and their proportion can reach 16% [57].
Going further, at the microscopic level, there are differences in the structure of the cardiomyocytes of patients with HFpEF compared to patients with HFrEF, with the cardiomyocytes of patients with HFpEF being thicker and less elongated [55]. However, according to Dao-Fu Dai et al., the fact that there is an age-dependent increase in the thickness of the left ventricular wall must be taken into account. This was shown by the analyses from the Framingham Heart Study and the Baltimore Longitudinal Study on Aging, studies that investigated apparently healthy adults using cardiac ultrasound. The conclusion was that there was an increased prevalence of left ventricular hypertrophy with age in both men and women, even in the absence of clinical hypertension, the most common risk factor for CVD [58]. And as aging is one of the most important contributors to HFpEF, left ventricular hypertrophy can be present but not necessarily in relation to HFpEF [58].
2.2. Left Ventricular Diastolic Dysfunction
The generally accepted definition of diastolic dysfunction states that the left ventricle is unable to fill to an adequate level, correlated to the body’s needs (end-diastolic volume—EDV) under conditions of low (but normal) pressure [59]. Although initially HFpEF was named as HF with diastolic dysfunction, diastolic dysfunction is not superimposed with HFpEF [59]. As Borlaug BA et al. state, diastolic dysfunction is independent of normal ejection fraction of the left ventricle. Diastolic dysfunction is the result of an abnormal distensibility of the LV that results in reduced relaxation and filling, regardless of whether or not the contractile function of the LV is normal, or whether or not these abnormalities produce symptoms [59]. It is accepted that diastolic dysfunction is part of normal human aging. The fact is reinforced by its detection in many people who do not have or will never have HFpEF. This occurs as a result of reduced filling of the LV in early diastole. This phenomenon becomes visible with increasing age in both sexes, through the decrease in ventricular elasticity due to the fibrosis of the LV walls and through the delay in active ventricular relaxation. Several mechanisms contribute to diastolic dysfunction like delayed relaxation due to reducing the efficiency of calcium capture and retention in the myocardial sarcoplasmic reticulum (SERCA2a), thus contributing to low sucking capability of the ventricle and wall rigidity. The development of diastolic dysfunction, in patients with HFpEF, does not affect the final filling volume of the LV, but this filling is difficult, as abnormally high filling pressures are required [55].
2.3. Ventricular Dyssynchrony
Ventricular dyssynchrony is defined as an increase in the time difference between the contractions of the two ventricles. The desynchronization of the moment of contraction of the two ventricles reduces the cardiac efficiency in performing the contraction and relaxation of the myocardium and can be correlated with the occurrence of HF [60]. It is known that cardiac dyssynchrony is, in the case of HFrEF, associated with a higher risk of adverse outcomes [61]. Although the electrical asynchrony in the case of HFpEF does not have as its main mechanism the bundle-branch block as in the case of HFrEF, these patients nevertheless tend to have wider QRS complexes, in these conditions, mechanical systolic and diastolic asynchrony being quite frequent [60,62]. To assess dyssynchrony, 2D speckle-tracking echocardiography (STE) is used to calculate global longitudinal strain. The advantage over the Doppler evaluation is its angle independence [63]. Patients with HFpEF have greater ventricular dyssynchrony compared to healthy people; this was expected, but dyssynchrony exists even in patients with a narrow QRS complex and LVEF ≥ 55% [62]. In HFpEF, mechanical dyssynchrony depends on QRS width (electrical dyssynchrony), ventricular hypertrophy, and diastolic but not systolic dysfunction [62].
2.4. Atrial Dysfunction and Atrial Fibrillation
Atrial fibrillation is a more frequent pathology in patients with HFpEF compared to those with HFrEF [64]. The left atrium (LA) functions as a buffer, absolutely necessary between the pulmonary veins and the LV. It is characterized by four functions. Two of them are passive and aim at blood storage, the reservoir function, respectively blood transfer into the LV, the conduit function. The other two are active, being represented by the accumulation of potential energy in the form of pressure, the battery function, respectively the contractile function that increases LV stroke output [65,66].
Studies have shown that once HFpEF is developed even at an early stage, patients need left atrium contraction to achieve normal LV filling compared to healthy individuals [65]. To maintain adequate LV filling, the atrial contribution, through contraction, becomes increasingly important with aging, but sustained atrial contraction also increases atrial pressure, which leads over time to atrial hypertrophy, which is a risk factor for atrial fibrillation [58]. The more important factor is the contractile function of the left atrium as patients with HFpEF, who develop atrial fibrillation, have a reduced quality of life due to reduced exercise capacity and the development or worsening of right ventricular (RV) dysfunction and increased mortality regardless of the severity of HF [67]. Melenovsky et al. found that the occurrence of atrial fibrillation in patients with HFpEF, compared to those in sinus rhythm, was associated with greater hemodynamic suffering of the right heart manifested by higher pressures in the pulmonary artery (PA), dilation and functional alteration of the right cavities [68].
2.5. Right Ventricle Dysfunction (RVD) and Pulmonary Vascular Disease
HfpEF is frequently associated with pulmonary hypertension (PH), right ventricular disease being part of this constellation of diseases included in the HfpEF syndrome. Up to two-thirds of patients with HfpEF present simultaneously PH [69]. Beyond the simple presence of PH, its severity is very important for a patient’s prognosis, as it is known that there is an increase in the relative risk of mortality of 28% for every 10 mmHg increase in PA pressure [70]. Reducing pulmonary arterial pressure, by using diuretics, decreases the number of hospitalizations for decompensated HFpEF [70].
RVD in most cases is caused by impaired pulmonary circulation, the most frequent cause being PH. Any increase in pulmonary arterial resistance requires an increase in myocardial contractility from the RV side, this increase being up to five times compared to the normal hemodynamic situation [71]. The problem is not that the afterload increases, but that it increases persistently, the RV not being able to cope with an increased pressure regime for long periods, given that the thickness of the RV wall is much reduced compared to that of the LV. As a result, the persistence of an increased arterial resistance will lead to RV dilation, decreased myocardial contractility (ventriculo-arterial decoupling), and decreased RV ejection fraction (RVEF). As a consequence, the RV cannot maintain an adequate cardiac output, resulting in the clinical appearance of HF.
Statistical data show that RVD can be found in up to 50% of patients with HFpEF, the development of RVD in patients with PH being a strong marker of increased morbidity and mortality, independent of the severity of HF [72].
2.6. Pericardial Restraint
The pericardial sac contributes to the good functioning of the heart through multiple roles, one of them being the limitation of the distension of the ventricular filling; this correlated with the venous return contributing to the increase in the intracardiac pressure [73]. Increased filling pressure in the LV is responsible for most of the symptoms in HFpEF. These elevated LV filling pressures occur in HFpEF predominantly due to myocardial relaxation abnormalities, but an important contribution also comes from pericardial constriction [74]. In an attempt to reduce intraventricular pressure, experiments were performed on animals in which it was demonstrated that pericardial resection reduces the increase in LV filling pressures in conditions of volume overload, both in normal hearts and those with diastolic dysfunction [73].
There are two phenotypes of HFpEF in which the pericardium contributes to the development and worsening of HF, the pulmonary hypertension phenotype and the obesity (cardiometabolic) phenotype [75,76]. In the case of patients with PH, it is understandable that the tension of the pericardial sac leads to an increase in RV pressure in exaggerated afterload conditions. In the obese phenotype, the combination of excess pericardial fat and increased cardiac volume leads to exaggeration of pericardial restraint [74].
2.7. Vascular Stiffness and Endothelial Dysfunction
Patients diagnosed with HFpEF frequently associate with reduced central aortic compliance and increased peripheral arterial stiffness [77]. This was expected as long as the increase in arterial stiffness is associated with diastolic dysfunction, with more arterial stiffness being responsible for the accelerated development of diastolic dysfunction [78]. Beyond the increased arterial stiffness, a sign of systemic vascular dysfunction, more than 70% of patients with HFpEF also associate with a functional coronary impairment manifested by a reduced coronary myocardial flow reserve [79]. The common element of the two vascular alterations (micro- and macro-vascular) is endothelial dysfunction. The same endothelial dysfunction was found in patients with HFpEF [80]. It is demonstrated that the presence and severity of endothelial dysfunction in patients with HFpEF contribute to a worse prognosis due to higher rates of acute cardiovascular events, a worse quality of life due to more severe symptoms, and reduced exercise capacity [77] (Figure 2).
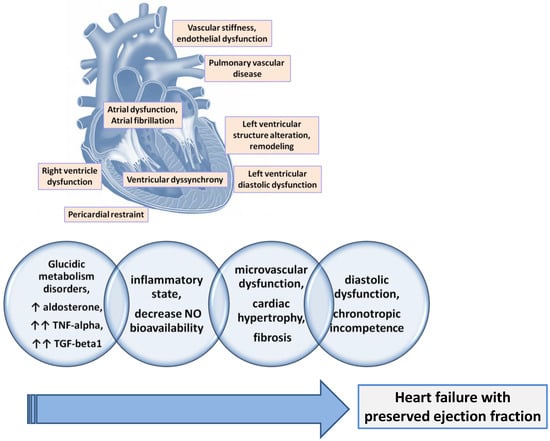
Figure 2. The main physiopathological mechanisms involved in the development of HFpEF.
The mechanism of endothelial dysfunction is the known one, mediated by the decrease in available nitric oxide (NO) by decreasing the activity of nitric oxide synthase 3 (NOS3) in endothelial cells or endothelial NO synthase (eNOS). But the reduction of NO bioavailability also leads to the reduction in soluble guanylate cyclase (sGC) activity in cardiomyocytes with direct negative consequences on cGMP production but also on protein kinase activity CGMP-dependent 1 (PRKG1) and, subsequently, titin phosphorylation, which leads finally to the stiffening of the cardiomyocytes. The trigger that affects NOS3 activity in patients with HFpEF is assumed to be the low-grade inflammation frequently found in their case [81] (Figure 2).
Recent research shows that cellular senescence is the basis of endothelial dysfunction. Cellular senescence is defined as the inability of the cell to divide. Beyond the natural senescence that occurs as a result of the shortening of telomeres, there is also premature senescence caused by inflammation and oxidative stress. On the other hand, senescent cells display a secretory status that in turn induces inflammation and senescence, thus creating a vicious circle [82,83]. The secretory phenotype associated with senescence consists in the secretion of growth factors (vascular endothelial growth factor), cytokines (IL-1 and IL-8), proteases (matrix metalloproteinase), and prothrombotic factors (plasminogen activator inhibitor 1) [82]. As a result, senescent cells promote tissue remodeling through cell proliferation associated with migration and tissue invasion. This tissue remodeling occurs under conditions of inflammation and oxidative stress. On the cardiovascular system, the effects consist in the development of atherosclerosis and ischemic coronary disease [84]. More recently, Andreas B. Gevaert and colleagues described the potential role of endothelial senescence in HFpEF using an accelerated senescence mouse model [85]. They found that exposure to a high-salt, high-fat diet accelerates endothelial senescence and promotes endothelial inflammation and endothelial dysfunction; this led to HFpEF as a result of the onset of diastolic dysfunction, left ventricular hypertrophy, left atrial dilatation, and interstitial fibrosis [85].
The mechanism by which endothelial dysfunction aggravates HFpEF seems to be related to the increase in central aortic pressure that prevents stroke volume from increasing, thus inducing the inability to reduce end-systolic volume in patients with HFpEF [86]. As a result of this fact (increased end-systolic volume), patients have a higher LV filling pressure at rest, and during exercise, a lower cardiac output reserve [87]. To support this theory, there are experimental data on animals that show that LV diastolic dysfunction (assessed by echocardiographic parameters) is limited if the nitroxyl donor, 1-nitrosocyclohexyl acetate (1-NCA), is administered chronically [88]. The supplementation of nitric oxide also led to a reduced level of the pro-fibrotic signal (Connective Tissue Growth Factor—CTGF) and to a reduced size of cardiomyocytes, elements that create the premise of reducing the stiffness of the LV wall [88]. Attempts to translate the data obtained on the animal model into clinical studies aimed at improving NO bioavailability and mitigating endothelial dysfunction have failed to demonstrate significant efficacy in patients with HFpEF. However, endothelial dysfunction and HFpEF share a series of elements (increased oxidative stress, inflammation, fibrosis) that cannot be ignored [9,89]. It remains to be seen how these experimental data will be exploited and transformed into therapeutic resources in clinical trials.
2.8. Chronotropic Reserve
Chronotropic incompetence (inability to increase heart rate) is a common functional alteration in patients with HFpEF, its prevalence varying between 57% and 77% depending on the study [86]. The cardiac output is directly influenced by the heart rate, it being calculated as the beat volume multiplied by the heart rate. One of the causes of exercise capacity limitations in patients with HFpEF is the inability to increase cardiac output appropriately due to the inability to increase heart rate during exercise. This fact is added to the impairment of the systolic volume reserve present in patients with HFpEF [87]. There are insufficient data to explain the mechanisms leading to the impairment of the chronotropic response in patients with HFpEF, but the assessment of chronotropic incompetence (CI) is important because it is a prognostic marker for increased risk of adverse clinical events [90]. Moreover, some studies showed a direct link between CI and reduction of peak oxygen consumption (VO2) in patients with HFpEF, with low VO2 being also associated with a worse prognosis in patients with HF [91]. One of the causes of CI appears to be impaired cardiac β-receptor sensitivity [92].
2.9. Cardiac Aging
Data from the literature show that one of the culprits responsible for the HFpEF epidemic is the aging of the general population, a process strongly associated with the development of diastolic dysfunction. Aging cannot be avoided, it is a normal process, but it involves cardiac and vascular alterations that are found often prematurely in patients with HFpEF. These alterations have already been mentioned here and are represented by diastolic dysfunction, chronotropic incompetence, loss of systolic reserve, endothelial dysfunction, and vascular stiffening [55]. They hide functional changes that occur at the cardiac and vascular levels as a result of aging. The functional changes aim at calcium metabolism with its deficient use in the myocytes, the decrease in β-adrenergic reserve, and the endothelial dysfunction related to the decrease in NO bioavailability. Physical deconditioning is added to these as a possible explanation or as a possible effect of them. The natural aging process of the heart can be accelerated by the presence of HFpEF [55], this unwanted contribution is greater in the case of women and overweight people [47].
The use of artificial intelligence (AI) in the analysis of ECG recordings made it possible to estimate the real age of the heart; this is greater than the chronological age (CA) in the case of premature cardiac senescence. This was possible using algorithms that measure PR interval, QRS duration, QT interval, and QTc interval [93]. Using this type of AI-supported algorithm, Frederik H. Verbrugge and colleagues demonstrated that premature cardiac senescence was greater in the obese [94]. This has been associated with obese patients with more frequent structural remodeling, more pronounced diastolic dysfunction, and a higher risk of atrial fibrillation [94]. Chenyu Li et al. linked obesity to inflammation as a possible mechanism for inducing cardiac senescence and HFpEF development [95]. They demonstrated the direct pro-inflammatory effects of nutrient overload. Nutrient overload can directly induce inflammation in cardiomyocytes, macrophages, and endothelial cells. Induction of inflammation seems to be based on “metabolic reprogramming”. Normally, cardiac macrophages help maintain homeostasis by removing senescent dead cells and defending against infections without inducing an immune response [96]. The metabolic reprogramming as a result of the surplus of nutrients causes macrophages to become promoters of inflammation with detrimental effects on the myocardium [95]. Metabolic reprogramming also takes place at the level of endothelial cells, this time as a result of inflammation; this reprogramming leads to the exacerbation of inflammation and the initiation of a vicious circle that leads to endothelial senescence and, in the end, endothelial dysfunction [97].
2.10. Hypervolemia
In HFpEF, there is no unanimous belief related to the presence of volume overload. Clinically, this overload is highlighted by the presence of edema, ascites, pleural collections, etc., signs absent most of the time in the case of HFpEF. Biologically, congestion is highlighted by measuring NPs. Even if these are increased, the magnitude of the increase is not great. In addition, their increase reflects ventricular parietal stress, not necessarily volume overload [98].
However, there is the general idea that, even in HFpEF, decompensation occurs due to congestion and volume overload, the kidneys being most likely responsible for this, patients with HFpEF presenting with a status of chronic cardio–renal suffering that involves the alteration of sodium and water homeostasis [98,99]. However, the way volume loading is presented differs between patients with HFrEF and those with HFpEF. If in those with HFrEF cardiac decompensation means more fluid in the vessels, in those with HFpEF it means more fluid in the interstitium without a significant increase in intravascular volume [100]. This fact can also explain the reduced effect of diuretics in patients with HFpEF compared to the significant effect of sodium–glucose co-transporter 2 inhibitors (iSGLT2) in improving dyspnea and reducing cardiac decompensation [100].
Judicious use of the terms “congestion” and “volume overload” is mandatory in order to use the right therapy, as congestion can occur even in the absence of volume overload [101].
This entry is adapted from the peer-reviewed paper 10.3390/ijms25020794
This entry is offline, you can click here to edit this entry!