Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
As an essential component of the innate immune system, the complement system is responsible for the defense against pathogens. The complement cascade has complex roles in the central nervous system (CNS), numerous reports have implicated the classical complement cascade in both brain development and decline. More specifically, complement dysfunction has been implicated in neurodegenerative disorders, such as Alzheimer’s disease (AD), which is the most common form of dementia. Synapse loss is one of the main pathological hallmarks of AD and correlates with memory impairment.
- complement system
- neurodegeneration
- Alzheimer’s disease
- neuroinflammation
- aging
- brain development
- pathogenesis
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia in the world and is characterized by a progressive loss of memory and other cognitive functions such as thinking and reasoning [1,2,3]. Currently, more than 55 million people worldwide are living with AD or related dementia, and this number is estimated to double every 20 years [2,4]. A better understanding of the disease mechanisms and development of efficient therapeutics are needed—both to combat the rising cost of healthcare associated with managing the disease and to address the trend towards a growing worldwide aging population which poses a risk factor for AD.
AD is characterized by two molecular pathological hallmarks: extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) [1]. The aggregation of Aβ plaques and NFTs is associated with significant neurodegeneration and synaptic loss as well as neuroinflammation [1]. Notably, synaptic loss is the strongest correlate for clinical dementia and more specifically for memory impairment in AD [5,6].
The literature in the AD field has historically described observations of altered neuroimmune function using vague terms such as neuroinflammation or glial “activation”, whereas several studies have now confirmed that alterations in immune cell phenotypes, gene expression, and morphology are complex processes with wide-ranging implications in human disease [7,8,9]. Interestingly, genome-wide association studies (GWAS) implicate glial cells in several neurodegenerative disorders, including AD [10,11,12]. The “traditional” concept of neuroinflammation has been modified in recent years, as a remarkable cellular heterogeneity is being noticed through the molecular profiling of the central nervous system (CNS) cell population and single-cell analysis [7,8,11,13].
The complement system is a central component of innate immunity that serves as a first line of defense against pathogens, eliminating them and removing both apoptotic cells and debris [14,15]. In the brain, the complement system serves a crucial role during brain development [16]. Within this context, separate proteins and pathways of complement have been described as key players in the formation, development, migration, and refinement of neurons [17,18,19]. In addition, there is a growing body of work, in both animal models and humans, suggesting that aberrant complement regulation may underlie several neurodegenerative diseases [20,21,22,23,24,25,26].
2. Complement System
As elucidated by Nonaka et al. [70], the complement cascade is an evolutionarily conserved system dating back more than a billion years. In 1895, complement was described as a heat-labile bactericidal component in serum [71]. Years later, one by one, the role of each essential component of the complement cascade was identified. The role of the complement system in innate immunity was discovered and elaborated on much later [72,73,74,75,76].
Currently, there are three well-defined complement-activation pathways: the classical, lectin, and alternative complement pathways (Figure 1). Notably, these pathways converge at C3 and downstream elements, which contribute to the formation of opsonins, anaphylatoxins, chemo-attractants, and the membrane attack complex (MAC) [77,78]. The complement system is regulated by both soluble proteins and cell surface proteins, which continually activate and inactivate these pathways to maintain homeostasis. When an immune reaction occurs against a pathogen, this system is amplified and mediates an inflammatory response that aids immune cells in fighting infection by enhancing (“complementing”) the ability of antibodies and phagocytic cells to remove microbes and damaged cells by targeting their plasma membrane.
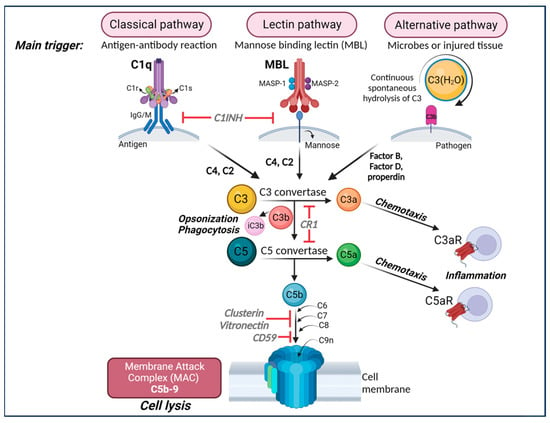
Figure 1. The complement cascade. The complement can be activated by three separate pathways: the classical, lectin, and alternative pathways. Cleavage of C3 is central to complement activation and its downstream effects lead to lysis and inflammatory signaling through the formation of the membrane attack complex (MAC). The classical pathway is activated upon binding of C1q to antigen–antibody complexes, consequently activating the two serine proteases C1r and C1s to form a complex with C1q known as the C1 complex. The C1 complex causes the cleavage of C4 and then C2, whose fragments combine to form C3 convertase (C4bC2a). The lectin pathway is initiated by binding carbohydrates such as mannose-binding protein (MBP) and ficolins on the surface of pathogens which activate two proteases, MASP1 and MASP2 (mannose-binding serine proteases 1 and 2), causing cleavage of C4 and then C2, whose fragments combine to form the same C3 convertase as in the classical pathway. The alternative pathway is always “on” at low levels in the blood and is further activated by pathogens including viruses, fungi, bacteria, LPS, etc., leading to the spontaneous hydrolysis of C3 and amplification of C3b, which binds Factor B and is cleaved by Factor D to form a different C3 convertase (C3bBb). Both C3 convertases cleave C3 into C3a and C3b. C3b can combine with the two different C3 convertases to form C5 convertase (C4bC2aC3b in the classical and lectin pathways and C3bBbC3b in the alternative pathway) which cleaves C5 into C5a and C5b. C3a and C5a are anaphylatoxins that bind their respective receptors, C3aR and C5aR, to recruit immune cells to sites of injury or infection which then secrete pro-inflammatory cytokines (Note: There are two forms of C5aR. C5aR1 is pro-inflammatory while C5aR2 is thought to be anti-inflammatory). Complement component C3b and its inactivated product, iC3b, opsonize or tag pathogens or immune complexes or weak synapses for phagocytic removal by microglia that bear their receptor, CR3. Complement component C5b binds C6, C7, C8, and finally multiple copies of C9 to form a channel (C5b-9 or MAC) in the cell membrane of a pathogen or cell, which induces lysis. Figure created with BioRender.com (accessed on 14 December 2023).
Most of the complement proteins are produced in the liver; however, it is progressively evident that complement proteins, as well as their receptors and regulators, are expressed throughout the CNS [79]. Although the functions of the complement cascade in the periphery are well deciphered, its functions in the CNS are less clear and under current investigation.
Several reports have shown that complement plays a critical role in the developmental processes of synaptic function and pruning [16,17,18,80,81] (Figure 2). In this process, inactive synapses are cleared to allow bolstering and maturation of more robust connections [17,18]. During the refinement of the visual system, for example, early components of the classical complement pathway localize to synapses, tagging them for removal by microglia, which express complement receptors, e.g., CR3/CD11b [16,17,18,51]. When this process occurs inadequately, brain connectivity is affected due to inappropriate connections. Interestingly, changes in brain connectivity have been described in several neurological disorders, including traumatic brain injury [25], multiple sclerosis [12,82,83], stroke, and AD [84,85]. The past decade studies have also demonstrated a role for complement in neurodevelopmental disorders such as autism and schizophrenia [86,87,88]. Significant findings have also revealed that complement components C3a, C3a receptor (C3aR), and C3 are critical in adult hippocampal neurogenesis [89,90]. Further, the role of complement in the pathogenesis of the Zika virus in the CNS has been shown in mouse models [91]. Recent reports have also highlighted the key role of complement activation in the development of neurological manifestations following SARS-CoV-2 infection [92,93]. Importantly, studies in both animal models and human brain tissue indicate that the impairment of complement regulation may contribute to AD pathogenesis [22,23,26,33,63,65,66,67].
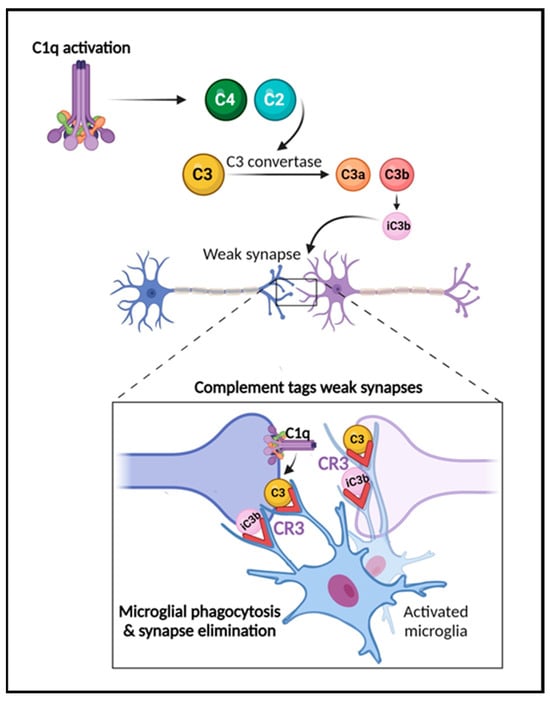
Figure 2. The role of the complement system in synapse elimination. The complement system serves a crucial role in normal brain development, aging, and AD progression by regulating synaptic pruning and elimination, as well as neuronal migration. The activation of the classical pathway leads to C1q expression causing cleavage of C4 and C2 and resulting in expression of C3 and iC3b. C3 and iC3b tag weak or unhealthy synapses representing an “eat me” signal. Microglia, which are responsible for maintaining homeostasis and surveilling the function of synapses, carry CR3 receptors (red) that recognize the tagged synapses and proceed with eliminating them via phagocytosis (synaptic engulfment). In aging and AD, this process has been linked to neurodegeneration. Figure created with BioRender.com (accessed on 14 December 2023).
3. Complement System in the Pathogenesis of AD
In the last 20 years, complement activation has been broadly studied in animal models in the context of mechanisms related to AD. Most AD mouse models express multiple AD-associated gene mutations and thereby emulate features of early-onset forms of AD. Some of these AD mouse models overexpress various human mutant APP genes that cause early onset, autosomal-dominant, familial AD either alone or in combination with one or more Presenilin-1 (PSEN1) autosomal dominant mutations that also lead to early onset familial AD. Newer knockin mouse models express physiological levels of mutant human APP. Overviews of these models are available on www.alzforum.org and in the following review [94]. Complement expression and its activation have been detected in AD brain tissue and mouse models [33,65,66,68,95,96]. It is now becoming increasingly clear that it serves a major role in AD pathology [16,21,23,97,98] (Figure 3). It has been shown that the classical complement proteins C1q, C3, and C4 co-localize with Aβ plaques and NFTs in memory-related areas in the AD brain [33,65,66,97,99]. Complement components and corresponding mRNA levels are also elevated in the AD brain and cerebrospinal fluid (CSF) [100,101]. Furthermore, increased levels of complement proteins have been reported in astrocyte-derived exosomes isolated from the plasma of AD patients [102]. A microarray study of young, healthy age, and AD brains demonstrated changes in complement-related gene expression in AD compared to age-matched controls [103]. GWAS have shown complement-related genes linked to AD [50,104,105]. Variants in the membrane protein complement receptor 1 (CR1) and the plasma regulator clusterin (CLU) genes of the complement pathway trigger late-onset AD, which is responsible for 95% of AD cases [104,106,107,108]. However, the mechanisms by which these complement genes affect AD pathology are still under investigation. Moreover, the role of complement in the inflammatory response observed in AD has also been widely explored in various reports [22,23,45,63,109,110,111].
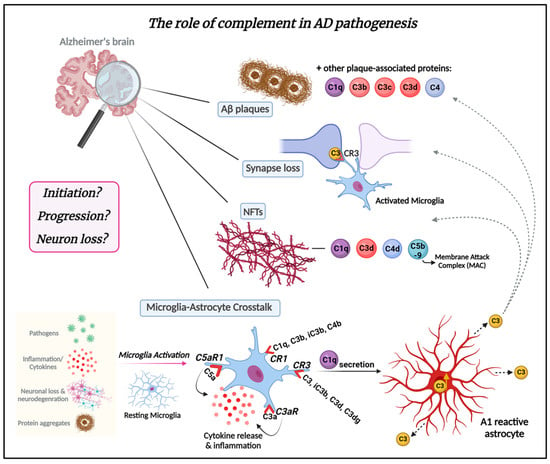
Figure 3. The role of the complement system in AD pathogenesis. The complement system plays a crucial role in AD pathogenesis. Complement proteins are deposited in amyloid plaques in Alzheimer’s brains and can be detected at early stages of plaque formation. Complement proteins C1q, C3b, C3c, and C3d are associated with amyloid plaques and dystrophic neurites therein throughout AD pathogenesis. Synapse loss is another critical feature of AD. Complement C3/C3b has emerged as a key player in synapse elimination in AD by tagging weak synapses and representing an “eat me” signal. Microglia carry CR3 receptors that recognize the tagged synapses and eliminate them via phagocytosis. Complement proteins, such as C1q, C3d, C4d, and C5b-9 also co-localize with neurofibrillary tangles (NFTs) in AD. The crosstalk between microglia and astrocytes is a significant part of AD pathogenesis and progression and may be mediated by complement. Microglia, the resident macrophages of the brain, can be activated in response to the presence of pathogens such as bacteria or viruses in the CNS, as well as by inflammatory signals (cytokines), signals released by injured or dying neurons, or in response to neurodegeneration. In addition, amyloid-β protein and tau aggregates can activate microglia. Once activated, microglia undergo morphological changes and release various molecules, including complement proteins such as C1q. Microglia express complement receptors CR1, CR3, C3aR, and C5aR1. CR1 binds complement opsonins C1q, C3b, iC3b, and C4b. CR3 binds C3 and its fragments C3d and C3dg, as well as iC3b (primary ligand). C3aR and C5aR1 (also known as CD88) specifically bind complement components C3a and C5a, respectively. The binding of these molecules to their corresponding receptors induces inflammation and cytokine release. C1q secreted by activated microglia induces A1-reactive astrocytes. A1 astrocytes are characterized by increased C3 expression and secretion. C3 released from A1-reactive astrocytes deposit on amyloid plaques, weakened synapses, and NFTs. It remains unknown whether complement is part of the initiation of AD pathology or drives the progression of the disease leading to neuron loss. Understanding the complex interplay between the complement system and AD is crucial for gaining insights into the molecular mechanisms driving AD and for the development of targeted therapeutic interventions. This knowledge not only holds the potential for addressing AD but also has broader implications for treating other neurodegenerative diseases. Figure created with BioRender.com (accessed on 4 January 2024).
C1q, the molecule that initiates the classical complement cascade, and C3, a downstream product and an “eat me” signal that attracts macrophages expressing C3 receptors (CR3), have been identified as key players in aging and AD pathogenesis [22,23,24,26,45,63,96,109,112]. Notably, fibrillar Aβ attaches to and activates C1q when immunoglobulins are not present, indicating that the classical complement cascade can be mobilized in AD independently of antibody-antigen immune complexes [28]. In the PS/APP mouse model, C1q co-localizes with fibrillar Aβ plaques [69]. In human AD postmortem brain tissue elevated C1q expression levels seem to positively correlate with Aβ plaques [113]. C1q has been demonstrated to localize with synapses and induce synaptic loss in AD mouse models [22] and has been suggested to act as an AD-specific modulator of the cellular crosstalk between microglia and astrocytes [114]. Recently, C1q was shown to be elevated in CSF-derived extracellular vesicles in AD patients compared to non-demented controls [115]. Furthermore, C1q has been shown to be increased in human and mouse brains with age [68,116]. On the other hand, deficiency of C1q in Tg2576 mice was shown to attenuate gliosis and synaptic degeneration without influencing Aβ levels [117]. Interestingly, deficiency of C1q in the 3xTg mouse model increased neurodegeneration [118]. These results suggest a critical role for the complement pathway in mediating AD pathology.
Recently, other complement components of synaptic dysfunction have been suggested to play a role in AD. Carpanini and colleagues showed that MAC fragments are located in the brains of mice in an APPNL-G-F AD-like mouse model [119]. Concomitantly, treatment with an anti-C7 antibody reduced synapse loss in aged mice [119]. Furthermore, deficiency of terminal pathway (C6-deficient mice) rescued synapse loss [119]. These data suggest that other complement components also drive synapse dysfunction in AD and open new avenues of therapies for neurodegenerative diseases.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25020817
This entry is offline, you can click here to edit this entry!