1. Cellular Chloroquine Effects
The major molecular mechanism believed to underly antitumor CQ and HCQ effects and make them potential tools for cancer therapy is their ability to suppress autophagy [
3,
15,
16]. Autophagy is an evolutionarily conserved intracellular process necessary for the maintenance of cellular homeostasis and the selective recycling of damaged proteins, macromolecular complexes, or whole organelles into lysosomes. Under conditions of nutrient deprivation or stress, autophagy is stimulated to supply the cells with an alternative energy source, thus promoting temporary survival [
20,
21]. A key process of autophagy is a transient generation of phagophores, sequestering structures that engulf unwanted cellular material and mature into double-membrane autophagosomes. Further fusion with lysosomes allows cargo degradation and turnover. The major molecular players of autophagy are Beclin-1, p62/SQSTM1 degrading scaffold protein, marker of autophagosomes LC3-II, and ATG proteins, which phosphorylate autophagy-related effectors and form the phagophores and autophagosomes.
Autophagy was implicated in the progression of cancers of different origins, with it at higher levels closely correlating with lower overall survival. However, its roles in these malignancies are complicated, as it can work as either a promoter or suppressor of cell death depending on the stage and type of cancer [
22,
23,
24]. By recycling the accumulated metabolites and positively regulating the metabolism of cancer cells, autophagy can function as a self-protective response against antitumor compounds, thus being a critical factor in the development of resistance to chemotherapy. On the other hand, recent studies indicate that a series of mutations such as RAS, BRAF, and p53 can alter the vulnerability of cancer cells to death and their sensitivity to cytotoxic drugs. Thus, chemotherapy-induced autophagy emerges as a promising critical target. It is believed that its suppression leads to the accumulation of autophagosomes, which can compromise cell viability and trigger apoptosis.
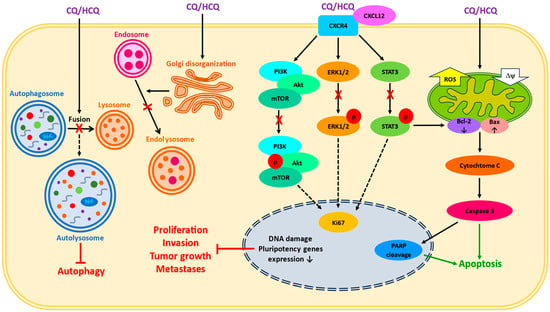
CQ and HCQ are lysosomotropic agents, which suppress the final step of autophagy by inhibiting the fusion of late endosomes with lysosomes (
Figure 1). After entering the cells, they passively diffuse into subcellular structures responsible for protein synthesis and recycling—Golgi vesicles, endosomes, and lysosomes. In acidic environments, they undergo protonation and remain trapped inside, thus causing alkalinization. This process inhibits the ability of enzymes to degrade unwanted material and blocks the survival mechanisms in cancer cells which allows them to proliferate [
3,
4].
Figure 1. A simplified scheme of reported CQ/HCQ effects in cancer cells. The impact on lysosomal and endosomal systems, the disturbances in intracellular signaling, and the induction of mitochondria-dependent apoptosis are presented. X—inhibition, ROS—reactive oxygen species, Δψ—mitochondrial membrane potential, p—phosphorylation.
2. Chloroquine as a Single Treatment
In many in vitro and in vivo studies, the application of CQ or HCQ as single agents has been found to effectively activate the cellular antitumor mechanisms, leading to both the induction of apoptosis and the suppression of autophagy. CQ inhibited the growth of orthotopic U87MG glioblastoma in a mouse model, whereas the decreased viability of cultured glioma cells was accompanied by the stimulation of caspase-3, pro-apoptotic protein Bax, and the p53 death pathway [
25]. Lakhter et al. [
26] showed that CQ reduces the growth of melanoma SKMe123 cells and mice melanoma xenografts via the lysosome-independent induction of apoptosis and prevention of PUMA protein degradation. The diminished tumorigenicity of primary pancreatic duct adenocarcinoma cells (PDAC) in the presence of CQ was a result of the inhibition of chemokine receptors CXCL12/CXCR4 and hedgehog signaling pathways accompanied by the downregulation of pluripotency-related genes. Such events led to the depletion of the cancer stem cells (CSCs) pool, although CQ had no effect on the growth of primary patient-derived pancreatic cancer xenografts in vivo [
27]. Moreover, CQ did not increase the LC3-II level in primary PDAC but inhibited autophagy in Panc1, 8988 T, and BxPC3 cell lines [
27]. The in vitro CQ treatment of liver HepG2 cancer cells resulted in G0/G1 cell cycle arrest, DNA damage, the activation of caspase-3 and pro-apoptotic protein Bim, PARP cleavage, and the loss of mitochondrial membrane potential, while an injection of CQ to mice bearing HepG2-GFP human liver cancer cells suppressed tumor growth [
28]. An addition of CQ to a pancreatic neuroendocrine neoplasm (PanNEN) culture induced ER stress and unfolded protein response via the activation of the PERK-eIF2α-ATF4 pathway, resulting in the expression of pro-apoptotic protein CHOP. In Men1 heterozygous-deficient (Men1
+/ΔN3-8) mice, a mouse PanNEN model, HCQ administration decreased tumor size and accelerated apoptosis, although proliferative activity was unchanged [
29]. In patient-derived glioblastoma stem cell lines with or without p53 mutations, CQ-suppressed proliferation was accompanied by the decreased activity of ATM (ataxia-telangiectasia mutated) and HIPK2 (homeodomain-interacting protein kinase
) kinases functioning as modulators of p53-mediated transcription [
30]. However, the survival of mice bearing glioblastoma xenografts following CQ administration greatly depended on p53 mutations [
30]. In human cervical cancer HeLa cells and osteosarcoma U2OS cells, CQ treatment induced the autophagy-independent disorganization of Golgi systems [
31]. The compromised mammosphere-forming efficiency of triple-negative breast cancer (TNBC) Hs578t, MDAMB231, and SUM159PT cells exposed to CQ in vitro and anti-metastasizing CQ effects in a mouse TNBC xenograft model were associated with a reduction in the tumorigenic CD44
+/CD24
−/low stem cell population accompanied by the inhibition of Jak2 and STAT3 phosphorylation, global DNA hypomethylation and damage, oxidative stress, mitochondrial membrane depolarization, and the release of cytochrome C to cytosol [
32,
33]. In a few cultured cell lines of adult T-cell leukemia/lymphoma (ATT) and a mouse Su9T01 tumor xenograft model, CQ or HCQ exerted a pronounced antitumor effect by rescuing the p47 protein, a negative regulator of the NF-κB pathway, from autophagy-lysosomal degradation, and via the downregulation of CADM1 (cell adhesion molecule 1) [
34].
The direct effect of CQ/HCQ on autophagy was confirmed in a series of other works. Thus, an increased number of autophagosomes and late endosomes, as well as an upregulation of LAMP, p62, and LC3-II proteins, have been reported in HeLa [
31], U2OS [
31], and TNBC cells [
32,
33]. The compromised proliferation and colony formation of endometrial adenocarcinoma cells with or without p53 mutations and an increased population of apoptotic cells after CQ treatment were also accompanied by the accumulation of autophagosomes, endosomes, LC3, and p62 [
35]. In human bladder cancer cell lines (RT4, 5637, and T24), CQ or HCQ inhibited proliferation and clonogenic formation via DNA fragmentation, increased apoptosis, the stimulation of caspases 3/7, PARP cleavage, the suppression of lysosome fusion, the accumulation of p62 and LC3-II [
36]. A similar inhibition of autophagy and stimulation of apoptosis was shown in brain [
30,
37], ovarian [
38], breast [
39,
40], thyroid [
41], and ATT [
34] cancer cells.
3. Chloroquine and Chemotherapy Drugs
3.1. Chloroquine and Doxorubicin (DOX)
Doxorubicin (DOX), a member of the Anthracyclines family, is widely used in chemotherapy against a variety of malignancies such as breast, genitourinary, and ovarian cancers; Hodgkin’s and non-Hodgkin’s lymphomas; Ewing and soft tissue sarcoma; lymphocytic and myelogenous leukemias; gastrointestinal, liver, and thyroid cancers; and neuroblastoma [
42,
43]. The molecular mechanisms of DOX's impact on cancer cells include intercalation into the DNA–topoisomerase II complex, which causes DNA damage, followed by p53-mediated cell cycle arrest, alterations in the redox state due to ROS accumulation and iron-dependent lipid peroxidation, the dysregulation of calcium-binding proteins and channels, and increased production of interleukins and interferons facilitating the immune-driven clearance of tumor cells. However, severe DOX cardiotoxicity leading to the death of cardiomyocytes and endothelial cells via autophagy, ferroptosis, necroptosis, or pyroptosis limits the benefits of DOX therapy [
44]. Moreover, long-term DOX therapy was reported to be associated with the development of resistance due to the activation of autophagy [
45,
46].
Combined applications of CQ or HCQ with DOX in in vitro and in vivo studies have confirmed the effectiveness of autophagy suppression in overcoming DOX resistance. In human hepatocellular carcinoma cells, an addition of a non-toxic CQ dose potentiated DOX cytotoxicity by diminishing its IC50 and preventing DOX-induced autophagy, evident from an increased LC3-II/LC3-I ratio and p62 expression [
47]. Co-treatment with CQ significantly sensitizes melanoma cells to DOX in vitro via the suppression of autophagy and enhancement of pyroptosis accompanied by the generation of the plasma membrane-targeting DFNA5-N fragment of gasdermin family protein DFNA5 [
48]. In cultured MCF-7 human breast cancer cells and the MCF-7 xenograft mouse model, CQ increased the sensitivity to DOX treatment and suppressed cell growth and aggressiveness via the downregulation of the Ki67 protein, a nuclear marker of active proliferation, the PPT1 enzyme involved in lysosomal degradation, and PI3K/Akt/mTOR signaling pathways [
39,
40,
49]. In TNBC HCC1806 cells, however, although DOX/CQ co-treatment reduced DOX doses and potentiated the growth inhibitory effect, such exposure also inhibited apoptosis, indicating the existence of alternative death pathways [
50]. Bano et al. [
51] showed an ability of CQ to enhance anticancer DOX effects in cervical cancer HeLa cells, where the synergistic effect was associated with the cleavage of procaspase-3 and PARP, upregulation of p62 and LC-3II, and decreased expression of LAMP-2, Syntaxin17, Rab5, and Rab7 proteins, which play a critical role in the fusion of autophagosomes with lysosomes. In human adenocarcinoma alveolar basal A549 cells, CQ accelerated DOX-induced apoptosis mediated by oxidative stress and led to the dephosphorylation of ERK kinases [
52]. DOX/CQ administered to mice inoculated with Ehrlich ascites carcinoma cells partially prevented the disruption of the alveolar structure, reduced the levels of antioxidant enzymes, and increased the level of neutrophil gelatinase-associated lipocalin (NGAL) playing an important role in bacterial defense and inflammation [
53]. Moreover, CQ therapy enhanced the anti-angiogenic effect of DOX in HUVECs [
54]. However, in thyroid cancer cell lines (TPC1, ACT1, and KTC1), CQ failed to enhance the efficacy of DOX [
41].
DOX/CQ was also tested in a series of new formulations proposed to decrease their doses and overcome prominent hydrophobicity [
3,
55,
56]. One such compound is PEGylated (poly(ethylene glycol)-coated) liposomal DOX (PLD) with a prolonged circulation time and increased microvascular permeability but without apparent cardiac toxicity [
42,
57]. A combination of CQ with PLD and pulse-wave ultrasound hyperthermia (pUH), a scheme developed to enhance the delivery of drugs to subcutaneous 4T1 breast cancer explant in BALB/c mice, induced the long-term suppression of tumor growth in comparison to CQ monotherapy or PLD + pUH treatment [
58,
59]. In HeLa cells, CQ enhanced the cytotoxicity of DOX encapsulated in pH-sensitive liposomes (SpHL-DOX) created to accelerate drug delivery in acidic environments [
60]. DOX/CQ co-loading in polyglycerol functionalized MoS
2 nanosheets (DOX/CQ-FPMoS
2) designed for targeted delivery and chemo-photothermal therapy enhanced the anticancer effect of laser irradiation in multidrug-resistant HeLa (HeLa-R) cells [
61]. The delivery of simultaneously encapsulated DOX⋅HCl and CQ in pH-responsive cholesteryl hemisuccinate self-assembled nanovesicles (DC-DIV/C) to DOX-resistant K562/ADR, and MCF-7/ADR cells or nude mice bearing a drug-resistant K562/ADR xenograft led to a much stronger antitumor effect accompanied by apoptosis and the blockage of autophagosome and lysosome fusion [
62].
3.2. Chloroquine and Paclitaxel (PTX)
Paclitaxel, a tricyclic diterpenoid belonging to taxanes and found in the bark and needles of
Taxus brevifolia, is one of the most successful natural chemotherapeutic compounds [
86,
87]. Due to minimal toxicity, high efficiency, and broad-spectrum antitumor activity, PTX is widely used for the therapy of ovarian, cervical, breast, colorectal, esophageal, lung, and prostate cancer, either alone or in combination with other agents. The major mechanism of its activity is a capacity to disrupt microtubule-assembling dynamics and induce cell cycle arrest at the G2/M phase, leading to apoptosis. However, as for other chemotherapeutic drugs, a major problem of PTX application is the development of chemoresistance due to protective autophagy [
88].
The synergic effects of CQ and PTX in suppressing viability and growth were accompanied by the inhibition of autophagy in MCF-7 human breast tumor cells [
39] and three TNBC cell lines [
32]. Moreover, CQ increased the sensitivity to PTX and reduced lung metastases, tumor growth, and recurrence in orthotopic murine MDAMB231 and SUM159PT tumor models and diminished the CD44
+/CD24
−/low CSC population in a clinical trial [
32]. The co-exposure of esophageal carcinoma EC109 cells to CQ and PTX was found to enhance the suppressive effect of PTX by inhibiting autophagy through the Akt/mTOR pathway [
89]. A phase II clinical trial, which recruited patients with advanced or metastatic breast cancer (of HR
+/HER2
− and TNBC types) who previously did not benefit from anthracycline-based chemotherapy, has shown that CQ in combination with taxane or taxane-like agents (paclitaxel, docetaxel, nanoparticle (NP) albumin-bound nab-paclitaxel, and ixabepilone) increases the objective response rate in comparison to that expected for PTX-based therapy itself, with good tolerance and a low rate of adverse effects [
90].
3.3. Chloroquine- and Platinum-Based Anticancer Drugs
The cohort of clinically approved platinating derivatives includes cisplatin (CIS), carboplatin (CPT), and oxaliplatin (OXP). The major mechanism of their action is DNA damage followed by the inhibition of transcription, but they are also able to exert cytoplasmic effects such as mitochondrial damage, ER stress, the suppression of ribosome biogenesis, and the elevation of micro-RNA activity [
91,
92]. They are widely used as a first-line chemotherapy compound for ovarian, cervical, testicular, bladder, esophageal, lung, and head and neck cancers; brain tumors; and neuroblastoma. However, the resistance and many side effects (nephrotoxicity, neurotoxicity, and hepatotoxicity) of these agents are reported, which drives the necessity to reduce their toxicity [
93].
Cisplatin. CQ enhanced the sensitivity to CIS treatment in endometrial adenocarcinoma cells [
35], thyroid cancer cell lines (TPC1, ACT1, and KTC1) [
41], and SH-SY5Y cells [
63]. In all of these cells, CQ effects were associated with the suppression of autophagy accompanied by increased LC3 and p62 expression. In epithelial ovarian cancer SKOV3 and hey cells, CQ alone had no effect on tumor migration and invasion capacities but alleviated CIS-induced autophagy with an upregulation of apoptosis-related proteins [
64]. In mice bearing a gastric cancer xenograft, CQ enhanced CIS chemosensitivity and the antitumor effect via the downregulation of multidrug resistance gene MDR1/P-gp and activation of caspase-3, as well as via the inhibition of CIS-triggered autophagy [
94]. In a mouse hepatocarcinoma xenograft model, CIS or CQ alone was able to reduce tumor growth; however, their combination significantly augmented the antitumor effect and impaired the proliferation of tumor cells by causing a higher level of apoptosis [
95]. The inhibition of autophagy with HCQ and CIS enhanced apoptosis and potentially therapeutic oxidative stress in neuroblastoma SH-SY5Y [
65].
Carboplatin. In combination with CPT, CQ exerted an additive antitumor effect in TNBC SUM159 stem cells and effectively reduced the growth of mice CPT-resistant SUM159 orthotopic xenografts proven to be linked with the inhibition of CPT-induced autophagy [
33]. The effectiveness of the CQ/CPT combination was confirmed in experiments on epithelial ovarian tumor cells from patients and mice xenografts, in which such a treatment decreased the CSCs pool, with surface co-expression of CD117 (c-Kit) and CD44, and suppressed their tumorigenic potential and spheroid-forming ability [
38]. In heavily pretreated patients with advanced solid tumors of different origin (GIST, neck and head, colorectal, urothelial, esophageal, etc.), a combination of CQ or HCQ with CPT increased progressive-free disease and overall survival (OS), although some side effects were reported [
96]. Importantly, in the exosomes obtained from the blood plasma of patients who received such treatment, both LC3-B isoforms were detected at advanced time points of the second and third cycles [
97].
Oxaliplatin. Apoptotic cell death induced by OXP was significantly enhanced by CQ treatment in hepatocellular carcinoma HepG2 cells with ATG7 knockdown due to the inhibition of autophagy [
66]. The application of CQ sensitized a few colon cancer cell lines to OXP under both oxic and hypoxic conditions and showed a synergistic interaction in suppressing the growth of mice HT29 xenografts with a reduced number of autophagosomal cells [
67]. Recently, biomimetic nanoparticles encapsulating both HCQ and OXP were shown to reduce the tumor capacities of hepatocellular carcinoma cells in vitro and in vivo by blocking or reversing autophagy [
68].
3.4. Chloroquine and Gemcitabine (GEM)
Gemcitabine is a nucleoside metabolic inhibitor whose active metabolites function as deoxycytidine analogs able to replace the building blocks of nucleic acids during DNA elongation, thus preventing DNA synthesis, arresting tumor growth, and promoting apoptosis [
98]. Although GEM was initially approved for the treatment of pancreatic cancer, it is currently used as an adjunct therapy for various solid tumors, such as ovarian cancer, non-small-cell lung carcinoma, and metastatic breast cancer. However, the resistance to GEM remains a serious problem among a noticeable rate of patients. It is not surprising that CQ was tested as a potential synergist to GEM.
In vivo CQ and GEM co-exposure more effectively eliminated tumors and improved the overall survival of mice bearing pancreatic patient-derived PDAC xenografts via the inhibition of the CXCL12/CXCR4 pathway with reduced phosphorylation of downstream effectors ERK and STAT3 and inhibition of hedgehog signaling [
27]. The addition of CQ strengthened the cytotoxic effects of GEM in human gallbladder cancer cells (GBCs) in vitro and inhibited the growth of GBC xenografts in mice in vivo, with an upregulation of the LC3-II/LC3-I ratio and Bax, downregulation of Bcl-2 and PARP, and inhibition of the Akt/mTOR pathway [
69]. The GEM/CQ combination significantly reduced the viability of human pancreatic cancer PANC-1 cells, although CQ alone did not exert any effect [
70]. The addition of CQ or HCQ to GEM therapy increased the OS of patients with advanced solid tumors of different types who previously received other treatment regimens [
96].
As for other chemotherapy drugs, new delivery strategies with enhanced penetration ability have been developed. The combined delivery of GEM and poly lactic-co-glycolic acid (PLGA) nanoparticles loaded with CQ, created as carriers to reduce its doses, to mice bearing orthotopic pancreatic cancer xenografts diminished tumor progression and suppressed the density of activated tumor cells at lower CQ doses [
99]. Chen et al. [
100] designed pH-sensitive PDGL-GEM@CAP/CQ particles consisting of GEM loaded in 6PA-modified DGL and co-precipitated with CQ and calcium phosphate. The administration of these particles to cultured pancreatic Pan 02 cells or mice bearing Pan 02 xenografts intensified antitumor GEM/CQ effects via the inhibition of proliferation, tumor growth, metastases and fibrosis, suppression of autophagy, and a decrease in the number of activated fibroblasts. In contrast to GEM monotherapy, adjuvant autophagy inhibition with HCQ significantly increased the median OS and DFS of the patients with high-risk PDAC [
101].
3.5. Chloroquine and Tyrosine Kinase Inhibitors
Imatinib (IMA). Imatinib is a small molecule tyrosine kinase inhibitor targeting numerous enzymes like CSF1R, c-KIT, FLT3, and platelet-derived growth factor receptor PDGFR-β, but it is reasonably selective to BCR-ABL fusion protein. It binds to the ATP pocket at a kinase active site, thus preventing the downstream phosphorylation of target proteins. IMA is the most common first-line cytotoxic agent for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GIST) in systemic therapy, but CML stem cells are intrinsically resistant to IMA [
105,
106].
An important role of autophagy in the resistance of CML cells to IMA was established in K562 cells, in which CQ or IMA alone did not change the rate of death while CQ/IMA co-treatment enhanced the sensitivity to IMA and accelerated apoptotic cell death. Moreover, the combination of these drugs produced the same effects in IMA-resistant lymphoid cell lines [
71]. CQ potentiated IMA-induced cytotoxicity and reduced the long-term viability of K562 cells due to the inhibition of autophagy initiation and autophagosome turnover [
72]. In GIST-T1 cells treated with CQ as a single agent or in combination with IMA, the suppressed growth and decreased viability were accompanied by increased LC3-II levels. Furthermore, treatment with IMA/CQ increased apoptosis in a mouse GIST-T1 xenograft [
73]. Although CQ or IMA alone did not inhibit or weakly inhibited the growth of GIST882 IMA-resistant cells, CQ addition enhanced the suppressive effect of IMA on cell proliferation and promoted apoptosis by blocking autophagy and altering the level of ERK phosphorylation [
74]. A phase II clinical trial, however, did not reveal any pronounced differences in long-lasting (12 and 24 months) “success” rates after 48-week administration of IMA/CQ, although the authors noticed some molecular responses [
107].
Lenvatinib. Lenvatinib is a potent tyrosine kinase inhibitor targeting PDGFRα, vascular endothelial growth factor receptors VEGFR1-3, fibroblast growth factor receptors FGFR1-4, tyrosine kinase receptor c-Kit, and RET proto-oncogene. It is widely used for the treatment of thyroid cancer and hepatocellular carcinoma [
108,
109]. Although the resistance and side effects following its application are common, data on Lenvatinib and CQ therapy are scarce. The effectiveness of CQ/Lenvatinib co-exposure was shown in thyroid cancer K1 and BCPAP cells, with the suppression of Lenvatinib-induced autophagy leading to the inhibition of proliferation and angiogenesis, increased apoptosis, and reduced VEGFA levels, while the co-treatment of mice bearing a K1 xenograft diminished tumor growth accompanied by decrease in VEGF markers VEGFA and CD31 and proliferation marker c-Myc [
75]. Combined HCQ/Lenvatinib therapy increased the overall survival of mice with hepatocellular carcinoma xenografts accompanied by the inhibition of tumor growth and lung metastases [
102].
Apatinib. Apatinib is a tyrosine kinase inhibitor that selectively inhibits VEGFR2 and has a mild activity towards c-Kit and c-SRC tyrosine kinases [
110]. The major anticancer effect of Apatinib is the blockage of angiogenesis, namely VEGF-mediated endothelial cell migration and proliferation leading to the suppression of new blood vessel formation in tumor tissue. The inhibition of Apatinib-induced autophagy with CQ in vitro increased apoptosis in thyroid cancer KHM-5M and C643 cells through the downregulation of p-Akt and p-mTOR, while Apatinib/CQ therapy augmented the suppression of the mice thyroid cancer xenograft in vivo [
76]. In ECA-109 and KYSE-150 esophageal squamous carcinoma cells, CQ administration enhanced the anticancer effects of Apatinib in vivo and in vitro by inhibiting autophagy via the IRE-1α–Akt–mTOR pathway and enhancing apoptosis via the stimulation of Bax and caspase-3 but decreasing the levels of Bcl-2 [
77].
3.6. Chloroquine and PI3K/Akt/mTOR Inhibitors
The PI3K/Akt/mTOR (phosphoinositide 3-kinase/Akt kinase/mammalian target of rapamycin) cascade is one of the most crucial signaling pathways controlling key cellular functions such as proliferation, growth, metabolism, and survival. Since its abnormal activation is a frequent event in many human malignancies, while the suppression leads to an upregulation of autophagy, the combination of PI3K/Akt/mTOR and autophagy inhibitors was suggested to have a higher therapeutic benefit [
111,
112,
113]. To date, more than 40 different agents targeting this pathway have been tested in various stages of clinical trials, but only a few of them have been approved for cancer therapy.
In MG63 osteosarcoma cells, CQ enhances apoptotic cell death promoted by mTOR inhibitor rapamycin (RAPA) by blocking the activity of downstream molecules of Akt/mTOR pathway 4E-BP1 and p70S6k, increasing the expression of autophagy-related proteins LC3-II and Atg12-Atg5, and decreasing the p62 level [
78]. Although CQ was not effective as a single treatment, CQ/RAPA exposure induced apoptosis via the overaccumulation of autophagosomes in well-differentiated human liposarcoma (WDLS) 93T449 cells [
79] and arrested the growth of dedifferentiated liposarcoma in mice bearing patient-derived orthotopic xenografts (DDLS PDOX) [
103].
The addition of CQ to Salidroside, a glycoside isolated from the root of
Rhodiola rosea L., enhanced the sensitivity of hepatocellular cancer HepG2 and 97H cells to this compound and exerted a synergic effect on the growth of the mice HepG2 xenograft by suppressing the invasion and metastasis of cancer cells through the PI3K/Akt/mTOR pathway, promoting mitochondrial dysfunction and altering the ratio between the expression of pro- and anti-apoptotic proteins [
80,
114]. The combination of imidazoquinoline derivative Dactolisib, dual PI3K/
mTOR inhibitor
, and Lys05, dimeric CQ compound, exerted a significant additive effect in the cultured lung cancer A549 cells via the stimulation of apoptotic genes, downregulation of proliferative gene marker
KI67, and blocking the expression of autophagic genes [
81]. In a few renal cancer cell lines, the synergic effects of CQ and Everolimus, RAPA analog approved for second-line therapy, included the suppression of cell viability, inhibition of autophagy, and shift to apoptosis via the intrinsic mitochondrial pathway associated with a decrease in the Beclin-1/Bcl-2 complex, although the tested cell lines had different sensitivities to such treatment [
82]. A phase I/II clinical trial that recruited patients with previously treated clear-cell renal carcinoma (ccRCC) showed that combined HCQ/Everolimus therapy is safe and tolerable and led to a partial response and prolonged stable disease in a subset of patients, although the mutations in the mTOR signaling pathway were associated with shorter survival [
115]. A significant antitumor capacity of HCQ combined with Temsirolimus, an intravenous RAPA analog, due to the modulation of autophagy was reported in a phase I clinical trial in patients with solid tumors and melanoma [
116].
3.7. Chloroquine and Other Agents
In PC-3 and LNCaP prostate cancer cell lines, combined treatment with the Palladium (Pd)(II) complex and CQ caused pyknotic nuclei and induced apoptosis accompanied by increased activity of caspase 3/7. Moreover, in PC-3 cells, such exposure downregulated autophagy proteins Atg5, Beclin-1, and LC3, pro-survival PI3K/Akt/mTOR-related protein, and Jak/STAT5, while p38 was highly phosphorylated [
83]. The study of Cook [
84] has shown that CQ addition augmented the sensitivity of breast cancer cells resistant to endocrine therapies to estrogen receptor-α (ERα)-targeted agents Tamoxifen or Faslodex both in vitro (in MCF7-RR, LCC9, and ZR-75-1/ICI-R cells) and in vivo (in mice xenograft models), with this effect linked with alterations in immune response. CQ supplementation inhibited autophagy and enhanced the cytotoxic effect of Sorafenib in TPC1, ACT1, and KTC1 thyroid cancer cell lines [
41]. The suppression of autophagy with CQ was able to improve the responses of the cultured brain tumor cells resistant to BRAF blockers to chemotherapy with MEK inhibitor Trametinib and, more importantly, reduce the metastases of brain glioblastoma in patients with BRAF mutations [
37]. HCQ enhanced apoptosis and potentially therapeutic oxidative stress in glioblastoma U-87 cells treated with Temozolomide, which possesses an ability to alkylate/methylate DNA, thus triggering its damage and the death of tumor cells [
65]. The combination of 5-FU with CQ significantly reduced the viability of a human pancreatic cancer PANC-1 cell line in comparison to a single 5-FU exposure, although CQ alone did not exert any effect [
70]. In a mouse xenograft hepatocarcinoma model, 5-FU or CQ alone was able to reduce tumor growth. However, their combination significantly augmented the antitumor effect and impaired the proliferation of tumor cells by causing a higher level of apoptosis [
95]. A few randomized clinical trials that attempted to use CQ as an adjuvant for conventional chemotherapy and radiotherapy of patients with glioblastomas (GBM) reported an enhanced response to antineoplastic treatment and improved mid-term survival [
117,
118]. A recent meta-analysis of clinical trials allowed the authors to conclude that CQ supplementation led to significantly improved survival or remission time and decreased mortality, with a low incidence of adverse effects and seizures, thus showing some effectiveness in the treatment of glioblastoma [
119]. A broad range of responses, from minor to partially good, and stable disease were reported in a study evaluating the effects of therapy with a combination of HCQ and Bortezomib, a reversible inhibitor of the chymotrypsin-like subunit of the 26S proteasome, in a group of patients with relapsed or refractory myeloma [
120].
3.8. Chloroquine in Multi-Drug Combinations
The development of chemoresistance and the existence of mutations have forced the search for new treatment combinations consisting of drugs acting on different cellular targets. In many of such combinations, CQ was added to suppress cytoprotective autophagy. In TNBC MDAMB231 or MDAMB468 cells, CQ potentiated the antitumor effect of the combined addition of PTX and PI3K/Akt/mTOR inhibitors Ipatasertib and Taselisib by reducing autophagic flux and enhancing apoptosis [
85]. In breast cancer MDAMB231 and MCF-7 cells, a triple combination of CQ, DOX, and Ixazomib, which binds the β5 subunit of the 20S proteasome, thus inhibiting its chymotrypsin-like activity
, synergistically suppressed cell growth and increased the sensitivity to chemotherapy [
121]. Using COAST (combination of autophagy selective therapeutics: CQ, Nelfinavir, RAPA, Dasatinib, and Metformin in 50% PEG400), Delaney et al. [
104] showed that this drug cocktail effectively arrested the growth of three types of mice xenografic ovarian cancers resistant to CIS-Docetaxel chemotherapy, with residual tumors exhibiting enhanced levels of LC3-II and ER stress marker GRP78. The combined addition of Apatinib and CQ enhanced the anti-proliferative effect of PTX on esophageal squamous carcinoma cells ECA-109 and KYSE-150 in vitro or intensified tumor suppression in vivo [
77]. A modest improvement in clinical responses (higher ORR and PFS) following combined HCQ/CPT/PTX therapy was reported in a study that recruited patients with newly diagnosed stage IV non-small-cell Kras-mutated lung cancer [
122]. Preoperative HCQ plus GEM/nab-PTX chemotherapy of the patients with potentially resectable pancreatic adenocarcinoma demonstrated an improved Evans grade histopathological response, decreased CA19-9 tumor marker level correlated with enhanced OS, and increased immune cell infiltration within the tumor [
123]. However, the addition of HCQ to conventional chemotherapy improved the histopathological response rate, but not OS, of patients suffering from PDAC with loss of tumor suppressor SMAD4 [
124] or patients with metastatic PDAC [
125].
4. Conclusions
Overall, the majority of experimental in vitro and in vivo works has shown that the addition of CQ or HCQ to conventional cytotoxic drugs significantly enhanced their anti-cancer effects, especially in cultured cells (Fig. 2). Therefore, these agents can be suggested as effective adjuvant agents sensitizing cancer cells to chemotherapy and offering more efficient elimination of tumors which can improve the clinically relevant curative rates. However, the clinical trials were not always successful, with the “partial response” being the most frequent finding. Some trials did not reveal any significant increase in overall surviving rates, probably due to enrollment of the patients with advanced stages of diseases or existence of undetected mutations. Another weakness of many clinical trials is the absence of control groups of patients, while the conclusions have been made based on “expected survival rate”. Moreover, long CQ and HCQ exposure is known to be associated with serious adverse effects such as allergic reaction, irreversible retinal toxicity, gastrointestinal discomfort, cardiomyopathy symptoms, neuromyotoxicity, and bone marrow suppression. Nevertheless, they should be further tested in experimental and clinical settings with the malignancies of different origin to reveal the types of tumors most sensitive to such treatment and the most effective chemotherapeutic combinations. Next, to more precisely target autophagy and diminish possible side effects, the development of new more specific and potent autophagy inhibitors is required.
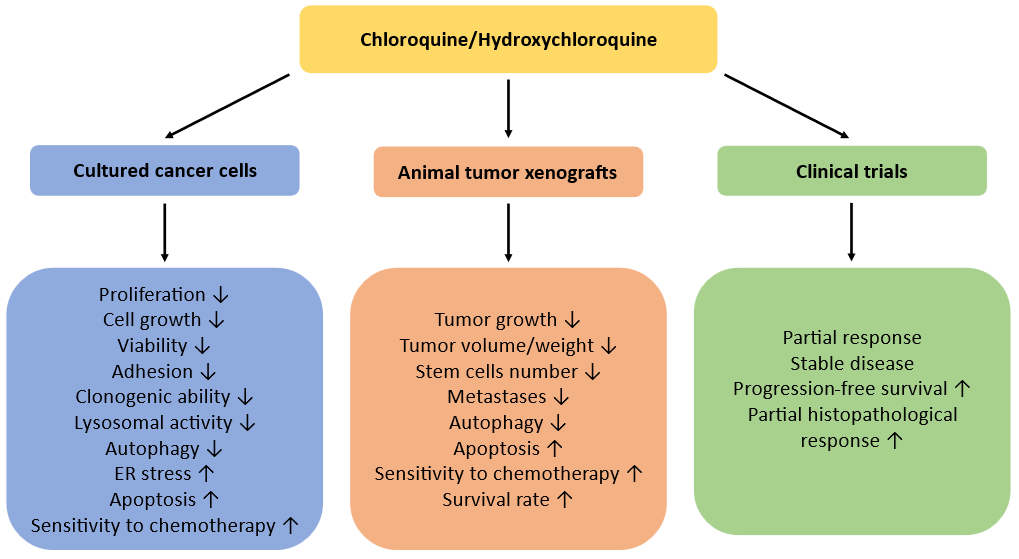
Fig. 2. Anticancer effects of CQ/HCQ in experimental studies and in clinical trials.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25020945