Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Multiple sclerosis (MS) is a chronic autoimmune inflammatory demyelinating disease of the central nervous system (CNS), which is triggered by an autoimmune assault targeting oligodendrocytes and myelin. Recent research indicates that the demise of oligodendrocytes due to an autoimmune attack contributes significantly to the pathogenesis of MS and its animal model experimental autoimmune encephalomyelitis (EAE).
- multiple sclerosis
- experimental autoimmune encephalomyelitis
- oligodendrocyte
- myelin
1. Introduction
Multiple sclerosis (MS) is an autoimmune inflammatory demyelinating disease of the central nervous system (CNS), driven primarily by T-cell-mediated inflammation [1][2][3][4]. The distinctive feature of MS pathology involves the occurrence of demyelinated plaques within the white matter of the CNS. These plaques are characterized by inflammation, the depletion of oligodendrocytes, demyelination, and the degeneration of axons. While the precise cause of MS remains uncertain, it is theorized to stem from an autoimmune assault targeting mature oligodendrocytes and the myelin sheath [1][2][3][4]. The prevailing theory suggests that the components of myelin induce the activation of T cells within the peripheral immune system of individuals with MS. Consequently, these T cells (with reactivity to myelin components) penetrate the blood–brain barrier, gaining entry into the CNS and instigating inflammation. In MS-related CNS inflammation, there is an infiltration of T cells, B cells, and monocytes, along with the activation of macrophages and microglia. This process leads to heightened levels of inflammatory cytokines and reactive oxygen species (ROS). The resulting inflammatory environment contributes to the demise of oligodendrocytes, myelin damage, and axon degeneration [1][2][3][4]. Genome-wide association studies (GWAS) have uncovered over 200 MS risk genes. A significant portion of these genes is linked to either autoimmunity or inflammation [1][2][3][4]. Experimental autoimmune encephalomyelitis (EAE) serves as the principal animal model for MS research, replicating numerous immunological, clinical, and pathological characteristics observed in MS [5][6][7][8]. In contrast to MS, which is a spontaneous and idiopathic disease, EAE is induced by inoculating animals, commonly mice and rats, with myelin components such as MOG (myelin oligodendrocyte glycoprotein), PLP (proteolipid protein), or MBP (myelin basic protein).
Oligodendrocytes play a crucial role in the CNS by producing the myelin sheath, which wraps around axons to provide insulation and protection [9][10]. In MS and EAE, oligodendrocytes face destruction from both specific, cell-selective immune mechanisms and non-specific bystander mechanisms [11][12][13]. Autoantibodies that target myelin components can trigger oligodendrocyte death and demyelination. This can occur through the activation of complement pathways or by binding to Fc-receptors on activated macrophages. Similarly, cytotoxic T cells, directed against myelin components, can induce oligodendrocyte death and demyelination. On the other hand, oligodendrocytes are particularly susceptible to ROS due to a combination of factors such as elevated intracellular iron, elevated metabolic rate, and reduced levels of antioxidant glutathione. Oxidative damage is a critical contributor to oligodendrocyte death in MS and EAE. Oligodendrocytes also express receptors that render them vulnerable to excitotoxic cell death. They carry kainate, AMPA, and NMDA receptors, making them susceptible to glutamate toxicity. Moreover, oligodendrocyte death can result from exposure to immune cytokines that are produced by myelin-reactive T cells. For instance, tumor necrosis factor α (TNFα) can cause oligodendrocyte apoptosis by binding to their p55 TNF receptor. Apart from these direct actions, immune cytokines have the potential to indirectly harm oligodendrocytes. This occurs through the activation of macrophages and microglia, leading to an upsurge in the production of ROS and inflammatory mediators [11][12][13].
Mounting evidence suggests that the death of oligodendrocytes, induced by autoimmune inflammation, significantly contributes to the development of MS and EAE [13][14]. The earliest structural alteration in newly developing demyelinating lesions in both MS and EAE is the apoptosis of oligodendrocytes, as observed in various studies [15][16]. Studies employing mice with an enforced expression of anti-apoptotic proteins (such as p35) or the knockout of pro-apoptotic proteins (such as the TNF receptor 1, Fas, and Fas-associated protein with death domain), specifically in oligodendrocytes, have demonstrated their protective effects on oligodendrocytes against inflammatory attacks. This protection leads to the amelioration of oligodendrocyte death, myelin damage, axon degeneration, and inflammation in the lesion sites and results in the attenuation of disease severity in the EAE model [17][18][19][20].
2. Mechanisms Regulating Oligodendrocyte Viability in MS and Its Animal Models
2.1. Immune Cytokines
Immune cytokines, including interferon-γ (IFN-γ) and TNFα, among others, are the key players in regulating the development of MS and EAE [1][2][3][4]. IFN-γ, a pleiotropic cytokine, intensifies inflammation by promoting the differentiation of Th1 T cells, activating microglia and macrophages, and inducing the expression of various inflammatory mediators. IFN-γ also influences oligodendrocyte viability in MS and its animal models [21][22][23]. IFN-γ applies its impact by attaching to its receptors on the cell surface, IFN-γR1 and IFN-γR2, triggering receptor oligomerization. This, in turn, activates Janus kinases 1 (JAK1) and JAK2, leading to the phosphorylation of both JAKs and the receptors [24][25]. Following receptor activation, the signal transducer and activator of transcription 1 (STAT1) is recruited and subsequently phosphorylated. Upon phosphorylation, STAT1 undergoes dimerization, moves to the nucleus, and regulates gene expression by attaching to gamma-activated sequence (GAS) elements found within the promoters of genes responsive to IFN-γ, including interferon regulatory factor 1 (IRF-1) [24][25]. On the other hand, suppressors of cytokine-signaling 1 (SOCS1) can inhibit IFN-γR-induced JAK/STAT1 signaling and modulate the impact of IFN-γ on cells [26].
IFN-γ, typically imperceptible in the healthy CNS, becomes detectable in the symptomatic phases of MS and EAE [22][27]. In vitro studies reveal that IFN-γ can directly act on oligodendrocytes to induce apoptosis [28][29]. However, the role of IFN-γ in MS and EAE is complex and sometimes contradictory. Administering IFN-γ to patients with MS and mice with EAE leads to enhanced inflammation in the CNS and exacerbated clinical symptoms [30][31][32]. Enforcing the expression of IFN-γ in the CNS induces inflammation and leads to abnormalities of oligodendrocytes and myelin in the CNS of transgenic mice [33][34]. Conversely, mice with a knockout of IFN-γ or its receptors maintain susceptibility to EAE, even developing the disease with increased morbidity and mortality [35][36]. Eliminating IFN-γ or its receptors renders mouse strains that are typically resistant to EAE susceptible to the disease [37]. CNS expression of IFN-γ before disease onset protects mice from EAE and prevents oligodendrocyte apoptosis and demyelination in the cuprizone model [38][39].
Furthermore, utilizing transgenic mice expressing SOCS1 exclusively in oligodendrocytes, a study demonstrated that enforced SOCS1 expression blocks oligodendrocyte response to IFN-γ, resulting in an acceleration of EAE development and an increase in EAE-induced oligodendrocyte apoptosis [40]. Intriguingly, GWAS has shown that IRF-1 is an MS risk gene [41]. IRF-1 knockout mice, while appearing normal under physiological conditions, exhibit resistance to EAE with abnormal IFN-γ responses [42]. Using transgenic mice expressing dominant-negative IRF-1 (dnIRF-1) exclusively in oligodendrocytes, a study showed that enforced expression of dnIRF-1 in oligodendrocytes attenuates disease severity and ameliorates oligodendrocyte apoptosis and myelin damage in the EAE model [43]. In summary, the available data collectively suggest that IFN-γ exerts its direct actions on oligodendrocytes in MS and EAE through IFN-γR-JAK/STAT-IRF-1 signaling, although its precise role in oligodendrocyte viability remains a subject of controversy.
2.2. Oxidative Stress
Oxidative stress stands as a prominent factor propelling tissue damage in inflammatory diseases, including MS and EAE [44][45]. This stress is primarily induced by the production of ROS, mainly by microglia and macrophages, during inflammation. Cells possess biological antioxidants, such as glutathione, ascorbic acid, and carotenoids, which interact with various oxidants to neutralize ROS and detoxify them. Nevertheless, when the production of ROS surpasses the cellular antioxidant capacity, elevated levels of ROS can induce the breakdown of essential cellular components, including lipids, proteins, and DNA, eventually culminating in cell apoptosis or necrosis [46][47]. Oligodendrocytes are highly susceptible to ROS because of reduced levels of antioxidant glutathione [48][49]. ROS can also disrupt the function of the mitochondrial respiratory chain, causing electron leakage and further contributing to oxidative injury [50]. Another factor that amplifies oxidative damage is the release of divalent iron from damaged cells into the extracellular space [51].
Cells possess an inherent mechanism designed to counteract excessive ROS and shield against oxidative damage. This mechanism, known as the oxidative stress response, is primarily regulated by the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) [52][53]. The modulation of Nrf2 activity primarily occurs at the protein level, regulated by its redox-sensitive inhibitor known as the kelch-like ECH-associated protein 1 (Keap1). In normal circumstances, Keap1 guides Nrf2 towards proteasomal degradation via polyubiquitination. However, when exposed to oxidants or electrophiles, crucial thiol groups within Keap1 undergo oxidation, leading to a conformational change in Keap1 and the subsequent liberation of Nrf2.
The level of Nrf2 is elevated in active MS lesions. The nuclear Nrf2 expression and upregulation of its downstream targets (such as heme oxygenase 1) are particularly strong in oligodendrocytes in MS lesions [54]. An in vitro study utilizing Nrf2 knockdown with shRNA demonstrated an exacerbation of oligodendrocyte apoptosis in response to oxidative stress. Conversely, Nrf2 activation achieved through the knockdown of its inhibitor, Keap1, showed an attenuation of oligodendrocyte apoptosis under oxidative stress conditions [55]. Several investigations have revealed that a global deficiency in Nrf2 exacerbates disease severity, demyelination, and inflammation in the EAE model [56][57]. On the other hand, compounds that activate Nrf2 signaling have been shown to reduce EAE disease severity. Notably, Dimethyl fumarate (DMF) induces modifications to thiol groups on the Nrf2 inhibitor Keap1, thereby stabilizing the Nrf2 protein and promoting the elevated expression of cytoprotective genes targeted by Nrf2, ultimately resulting in the attenuation of EAE disease severity [58][59][60].
2.3. Mitochondrial Damage
Mitochondria, which are double-membrane-bound subcellular organelles, are vital for fundamental cellular processes such as ATP production, calcium signaling, and iron homeostasis [61][62][63]. They face constant challenges from oxidative stress due to ROS generated in the electron transport chain [61][62][63][64]. Maintaining a proper mitochondrial function relies on a complex web of mitochondrial quality-control mechanisms, encompassing ROS scavenging, DNA repair, protein refolding/degradation, mitochondrial fusion and fission, mitophagy, and mitochondrial biogenesis [61][62][63][64]. Mitochondrial fusion promotes the mixing of content between healthy and partially dysfunctional mitochondria, while fission segregates damaged components [63][65]. Mitophagy selectively targets damaged or dysfunctional mitochondria for lysosomal degradation [63][66].
Mitochondrial dysfunction/damage in oligodendrocytes has been implicated in various demyelinating diseases [67][68][69]. Oligodendrocyte death and demyelination are observed in various inherited mitochondrial diseases in humans, such as Leber’s hereditary optic neuropathy (LHON) and Kearns–Sayre Syndrome [69][70]. LHON mutations also increase the risk of developing MS [71]. A study showed that double-strand breaks of mitochondrial DNA (mtDNA) in oligodendrocytes result in mitochondrial dysfunction, oligodendrocyte apoptosis, and demyelination in mice [72]. Data indicate that neurotoxin cuprizone causes oligodendrocyte apoptosis in the cuprizone-induced demyelination model by inducing mitochondrial dysfunction [73][74][75]. Notably, evidence suggests that inflammation-induced mitochondrial damage contributes to oligodendrocyte apoptosis in MS and EAE [67][68][69][76][77][78].
Furthermore, several studies suggest that mitophagy influences oligodendrocyte viability in demyelinating diseases. However, these studies are highly contradictory [78][79]. It has been shown that the concentrations of PARKIN and PINK1 (the master regulators of mitophagy) are elevated in the serum and cerebrospinal fluid of patients with MS [80][81].
2.4. The UPR
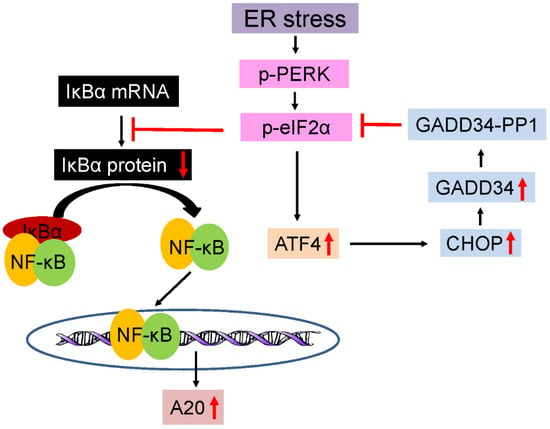
The endoplasmic reticulum (ER) serves as the hub for the modification and folding of membrane and secretory proteins in eukaryotic cells. Disruptions in protein modification or folding cause the buildup of unfolded or misfolded proteins within the ER, resulting in ER stress and the activation of ER stress sensors, including inositol requiring enzyme 1 (IRE1), pancreatic ER kinase (PERK), and activating transcription factor 6 (ATF6). The activation of these three ER stress sensors coordinates an adaptive program known as the UPR [82][83][84]. The activation of PERK inhibits global protein translation but promotes the translation of the transcription factor ATF4 by phosphorylating eukaryotic translation initiation factor 2α (eIF2α). ATF4, in turn, enhances the expression of various stress-responsive genes, including cytoprotective genes and CHOP (CAATT enhancer–binding protein homologous protein). The upregulation of CHOP downregulates the activity of the PERK-eIF2α pathway by increasing the expression of GADD34 (growth arrest and DNA damage 34), which, along with PP1 (protein phosphatase 1), dephosphorylates p-eIF2α (phosphorylated eIF2α), establishing a robust negative feedback loop (Figure 1).
Figure 1. Schematic diagram of the PERK-eIF2α-NF-κB-A20 pathway. ER stress initiates PERK phosphorylation, leading to phosphorylation of eIF2α. P-eIF2α facilitates the translation of ATF4. ATF4, in turn, stimulates the expression of CHOP. CHOP induction downregulates the PERK-eIF2α pathway by increasing the expression of GADD34, which forms a complex with PP1, known as the GADD34-PP1 complex, to dephosphorylate p-eIF2α. P-eIF2α also triggers the activation of NF-κB pathway by inhibiting the translation of IκBα. NF-κB activation can stimulate the expression of multiple anti-apoptotic proteins, including A20/TNFAIP3.
2.5. NF-κB Signaling
The transcription factor NF-κB plays a critical role in modulating inflammation and cell viability in inflammatory diseases like MS and EAE [85][86][87]. It manifests as a heterodimer or homodimer within the Rel family, encompassing p65, c-Rel, RelB, p50, and p52 [88][89]. In its inactive state, NF-κB is confined to the cytoplasm by interacting with its inhibitors (IκBs). Upon activation, NF-κB separates from IκBs and relocates to the nucleus, where it binds to the κB consensus DNA sequence, triggering the transcription of genes associated with inflammation and cell viability. NF-κB can be activated by various pathways, including the IκB kinase 2 (IKK2)-dependent canonical pathway, the noncanonical pathway, and atypical pathways [85][89][90]. The canonical pathway is triggered when cell surface receptors bind to their ligands (such as TNF receptors binding with TNFα), leading to the formation of the IKK complex, composed of IKK1, IKK2, and NEMO.
The NF-κB pathway is activated in both inflammatory cells and oligodendrocytes in MS and EAE [16][91][92]. While it is well-documented that the activation of the NF-κB pathway in inflammatory cells, such as T cells and monocytes, contributes to the development of MS and EAE by fostering inflammation [85][87][93][94][95], there is intriguing evidence suggesting a protective role for NF-κB activation in oligodendrocytes against inflammation in these conditions [85].
Evidence suggests that the IKK2-dependent canonical pathway can activate NF-κB in oligodendrocytes in MS and EAE [87][96]. Recent studies demonstrated that the activation of the PERK-eIF2α pathway in response to ER stress triggers the activation of NF-κB signaling in oligodendrocytes by repressing the translation of IκBα (an atypical pathway) (Figure 1) [16][97][98].
NF-κB is recognized for its cytoprotective role by inducing specific anti-apoptotic genes, such as A20/TNFAIP3, cIAPs, cFLIP, Bcl-2, and/or Bcl-xL (Figure 1) [89][99][100]. However, the precise mechanisms through which NF-κB activation safeguards oligodendrocytes against inflammation in MS and EAE remain elusive. A recent study, employing whole-genome RNA sequencing, revealed that mild NF-κB activation induced by IKKca expression, specifically in oligodendrocytes, significantly upregulated only 12 genes. Among these, A20/TNFAIP3, an NF-κB-target, anti-apoptotic gene, stood out [97].
3. Therapeutic Potential and Future Directions
Growing evidence indicates that the death of oligodendrocytes caused by inflammation significantly contributes to the development of MS. To impede disease progression in MS patients, it is imperative to explore therapeutic approaches that safeguard oligodendrocytes from inflammation. Despite notable advancements in anti-inflammatory treatments for MS, there remains a critical gap in the absence of an effective intervention to mitigate oligodendrocyte death and demyelination. A primary hurdle in MS research involves comprehending the mechanisms dictating oligodendrocyte viability and formulating therapeutic strategies to shield these cells and preserve myelin in the face of inflammation.
Accumulating evidence suggests that targeting the Nrf2-mediated oxidative stress response holds promise as a therapeutic strategy for a range of diseases, including MS [101][102]. Several small chemical compounds that selectively influence the activity of the Nrf2-mediated oxidative stress response have been identified, including DMF, SF (isothiocyanate), CDDO-Me (triterpenoid), and RTA 408 (triterpenoid). Several of these compounds have advanced to clinical trials for treating various diseases. Notably, DMF has received approval for use in the treatment of MS [101][102][103]. Significant advancements have been achieved in the identification of small chemical compounds that specifically modulate the activity of the three individual pathways of the UPR [104][105].
The current available data highlight the crucial roles played by IFN-γ signaling, oxidative stress, mitochondrial damage, the UPR, and NF-κB signaling in influencing oligodendrocyte viability in MS. However, their precise roles and underlying mechanisms remain unclear and necessitate further investigation. Conversely, it is established that oligodendrocyte progenitor cells (OPCs) possess the ability to proliferate and differentiate into remyelinating oligodendrocytes responsible for repairing myelin damage in demyelinated lesions in MS. Despite this regenerative potential, MS exhibits insufficient oligodendrocyte regeneration and remyelination, leading to the accumulation of unrepaired lesions and a progressive decline in neurological function [106][107][108][109].
This entry is adapted from the peer-reviewed paper 10.3390/cells13020116
References
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952.
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955.
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180.
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43.
- Steinman, L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron 1999, 24, 511–514.
- Kipp, M.; Vander, S.B.; Vogel, D.Y.; Puentes, F.; Valk, P.; Baker, D.; Amor, S. Experimental in vivo and in vitro models of multiple sclerosis: EAE and beyond. Mult. Scler. Relat. Disord. 2012, 1, 15–28.
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain. Pathol. 2017, 27, 123–137.
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244.
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927.
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431.
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53.
- Patel, J.; Balabanov, R. Molecular mechanisms of oligodendrocyte injury in multiple sclerosis and experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 2012, 13, 10647–10659.
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14, 599717.
- Prineas, J.W.; Parratt, J.D. Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 2012, 72, 18–31.
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004, 55, 458–468.
- Lin, W.; Lin, Y.; Li, J.; Fenstermaker, A.G.; Way, S.W.; Clayton, B.; Jamison, S.; Harding, H.P.; Ron, D.; Popko, B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 2013, 33, 5980–5991.
- Hisahara, S.; Araki, T.; Sugiyama, F.; Yagami, K.; Suzuki, M.; Abe, K. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J. 2000, 19, 341–348.
- Hisahara, S.; Okano, H.; Miura, M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci. Res. 2003, 46, 387–397.
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F. Apoptosis of Oligodendrocytes via Fas and TNF-R1 Is a Key Event in the Induction of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 175, 5875–5884.
- McGuire, C.; Volckaert, T.; Wolke, U.; Sze, M.; de Rycke, R.; Waisman, A. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J. Immunol. 2010, 185, 7646–7653.
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189.
- Popko, B.; Corbin, J.G.; Baerwald, K.D.; Dupree, J.; Garcia, A.M. The effects of interferon-gamma on the central nervous system. Mol. Neurobiol. 1997, 14, 19–35.
- Popko, B.; Baerwald, K.D. Oligodendroglial response to the immune cytokine interferon gamma. Neurochem. Res. 1999, 24, 331–338.
- Gysemans, C.; Callewaert, H.; Overbergh, L.; Mathieu, C. Cytokine signalling in the beta-cell: A dual role for IFNgamma. Biochem. Soc. Trans. 2008, 36, 328–333.
- Ng, C.T.; Fong, L.Y.; Abdullah, M.H. Interferon-gamma (IFN-γ): Reviewing its mechanisms and signaling pathways on the regulation of endothelial barrier function. Cytokine 2023, 166, 156208.
- Krebs, D.L.; Hilton, D.J. SOCS: Physiological suppressors of cytokine signaling. J. Cell. Sci. 2000, 113 Pt 16, 2813–2819.
- Imitola, J.; Chitnis, T.; Khoury, S.J. Cytokines in multiple sclerosis: From bench to bedside. Pharmacol. Ther. 2005, 106, 163–177.
- Baerwald, K.D.; Popko, B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J. Neurosci. Res. 1998, 52, 230–239.
- Lin, W.; Harding, H.P.; Ron, D.; Popko, B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell. Biol. 2005, 169, 603–612.
- Panitch, H.S.; Hirsch, R.L.; Schindler, J.; Johnson, K.P. Treatment of multiple sclerosis with gamma interferon: Exacerbations associated with activation of the immune system. Neurology 1987, 37, 1097–1102.
- Renno, T. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol. Cell. Neurosci. 1998, 12, 376–389.
- Sun, D.; Newman, T.A.; Perry, V.H.; Weller, R.O. Cytokine-induced enhancement of autoimmune inflammation in the brain and spinal cord: Implications for multiple sclerosis. Neuropathol. Appl. Neurobiol. 2004, 30, 374–384.
- Corbin, J.G. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol. Cell. Neurosci. 1996, 7, 354–370.
- LaFerla, F.M.; Sugarman, M.C.; Lane, T.E.; Leissring, M.A. Regional hypomyelination and dysplasia in transgenic mice with astrocyte-directed expression of interferon-gamma. J. Mol. Neurosci. 2000, 15, 45–59.
- Ferber, I.A. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 1996, 156, 5–7.
- Willenborg, D.O.; Fordham, S.; Bernard, C.C.; Cowden, W.B.; Ramshaw, I.A. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J. Immunol. 1996, 157, 3223–3227.
- Krakowski, M.; Owens, T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 1996, 26, 1641–1646.
- Furlan, R. Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J. Immunol. 2001, 167, 1821–1829.
- Gao, X.; Gillig, T.A.; Ye, P.; D’Ercole, A.J.; Matsushima, G.K.; Popko, B. Interferon-gamma protects against cuprizone-induced demyelination. Mol. Cell. Neurosci. 2000, 16, 338–349.
- Balabanov, R.; Strand, K.; Goswami, R.; McMahon, E.; Begolka, W.; Miller, S.D. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 2013–2024.
- Fortunato, G.; Calcagno, G.; Bresciamorra, V.; Salvatore, E.; Filla, A.; Capone, S.; Liguori, R.; Borelli, S.; Gentile, I.; Borrelli, F.; et al. Multiple sclerosis and hepatitis C virus infection are associated with single nucleotide polymorphisms in interferon pathway genes. J. Interferon. Cytokine. Res. 2008, 28, 141–152.
- Matsuyama, T.; Kimura, T.; Kitagawa, M.; Pfeffer, K.; Kawakami, T.; Watanabe, N.; Kundig, T.M.; Amakawa, R.; Kishihara, K.; Wakeham, A. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lym-phocyte development. Cell 1993, 75, 83–97.
- Ren, Z.; Wang, Y.; Tao, D.; Liebenson, D.; Liggett, T.; Goswami, R. Overexpression of the dominant-negative form of interferon regulatory factor 1 in oligodendrocytes protects against experimental autoimmune encephalomyelitis. J. Neurosci. 2011, 31, 8329–8341.
- Tobore, T.O. Oxidative/Nitroxidative Stress and Multiple Sclerosis. J. Mol. Neurosci. 2021, 71, 506–514.
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67.
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163.
- Klein, J.A. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Invest. 2003, 111, 785–793.
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022.
- Juurlink, B.H.; Thorburne, S.K.; Hertz, L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia 1998, 22, 371–378.
- Lassmann, H.; Van, H.J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510.
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468.
- Zhang, H.; Davies, K.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free. Radic. Biol. Med. 2015, 88 Pt B, 314–336.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474.
- Licht-Mayer, S.; Wimmer, I.; Traffehn, S.; Metz, I.; Brück, W.; Bauer, J.; Bradl, M.; Lassmann, H. Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta Neuropathol. 2015, 130, 263–277.
- Liessem-Schmitz, A.; Teske, N.; Scheld, M.; Nyamoya, S.; Zendedel, A.; Beyer, C.; Clarner, T.; Fragoulis, A. Nrf2 Signaling in Sodium Azide-Treated Oligodendrocytes Restores Mitochondrial Functions. J. Mol. Neurosci. 2018, 66, 229–237.
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513.
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246.
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: Therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803.
- Kasarełło, K.; Jesion, A.; Tyszkowska, K.; Matusik, K.; Czarzasta, K.; Wrzesień, R.; Cudnoch-Jedrzejewska, A. Effect of dimethyl fumarate on heme oxygenase-1 expression in experimental allergic encephalomyelitis in rats. Folia. Neuropathol. 2017, 55, 325–332.
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30.
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562.
- Whitley, B.N.; Engelhart, E.A.; Hoppins, S. Mitochondrial dynamics and their potential as a therapeutic target. Mitochondrion 2019, 49, 269–283.
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell. 2021, 56, 881–905.
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1066–1077.
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53.
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell. Biol. 2020, 219, e202004029.
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell. Mol. Biol. 2017, 328, 49–103.
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 25, 334–343.
- Carvalho, K.S. Mitochondrial dysfunction in demyelinating diseases. Carvalho KS. Semin. Pediatr. Neurol. 2013, 20, 194–201.
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155.
- Vanopdenbosch, L.; Dubois, B.; D’Hooghe, M.B.; Meire, F.; Carton, H. Mitochondrial mutations of Leber’s hereditary optic neuropathy: A risk factor for multiple sclerosis. J. Neurol. 2000, 247, 535–543.
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. 2017, 37, 10185–10199.
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain. Pathol. 2001, 11, 107–116.
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505.
- Vega-Riquer, J.M.; Mendez-Victoriano, G.; Morales-Luckie, R.A.; Gonzalez-Perez, O. Five Decades of Cuprizone, an Updated Model to Replicate Demyelinating Diseases. Curr. Neuropharmacol. 2019, 17, 129–141.
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735.
- Steudler, J.; Ecott, T.; Ivan, D.C.; Bouillet, E.; Walthert, S.; Berve, K.; Dick, T.P.; Engelhardt, B.; Locatelli, G. Autoimmune neuroinflammation triggers mitochondrial oxidation in oligodendrocytes. Glia 2022, 70, 2045–2061.
- Wang, M.R.; Zhang, X.J.; Liu, H.C.; Ma, W.D.; Zhang, M.L.; Zhang, Y.; Li, X.; Dou, M.M.; Jing, Y.L.; Chu, Y.J.; et al. Matrine protects oligodendrocytes by inhibiting their apoptosis and enhancing mitochondrial autophagy. Brain. Res. Bull. 2019, 153, 30–38.
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118.
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131.
- Cossu, D.; Yokoyama, K.; Sechi, L.A.; Hattori, N. Potential of PINK1 and PARKIN Proteins as Biomarkers for Active Multiple Sclerosis: A Japanese Cohort Study. Front. Immunol. 2021, 12, 681386.
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell. 2022, 82, 1477–1491.
- Yue, Y.; Stone, S.; Lin, W. Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural. Regen. Res. 2018, 13, 1507–1515.
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect Biol. 2009, 1, a001651.
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-κB) in multiple sclerosis pathology. Trends. Mol. Med. 2013, 19, 604–613.
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362.
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234.
- Mincheva-Tasheva, S.; Soler, R.M. NF-κB signaling pathways: Role in nervous system physiology and pathology. Neuroscientist 2013, 19, 175–194.
- Bonetti, B.; Stegagno, C.; Cannella, B.; Rizzuto, N.; Moretto, G.; Raine, C.S. Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am. J. Pathol. 1999, 155, 1433–1438.
- Gveric, D.; Kaltschmidt, C.; Cuzner, M.L.; Newcombe, J. Transcription factor NF-kappaB and inhibitor I kappaBalpha are localized in macrophages in active multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 1998, 57, 168–178.
- Greve, B.; Weissert, R.; Hamdi, N.; Bettelli, E.; Sobel, R.A.; Coyle, A.; Kuchroo, V.K.; Rajewsky, K.; Schmidt-Supprian, M. I kappa B kinase 2/beta deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 179–185.
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640.
- Hao, W.; Decker, Y.; Schnöder, L.; Schottek, A.; Li, D.; Menger, M.D.; Fassbender, K.; Liu, Y. Deficiency of IκB Kinase β in Myeloid Cells Reduces Severity of Experimental Autoimmune Encephalomyelitis. Am. J. Pathol. 2016, 186, 1245–1257.
- Yan, J.; Greer, J.M. NF-kappa B, a potential therapeutic target for the treatment of multiple sclerosis. CNS. Neurol. Disord. Drug. Targets. 2008, 7, 536–557.
- Lei, Z.; Yue, Y.; Stone, S.; Wu, S.; Lin, W. NF-κB Activation Accounts for the Cytoprotective Effects of PERK Activation on Oligodendrocytes during EAE. J. Neurosci. 2020, 40, 6444–6456.
- Lin, Y.; Jamison, S.; Lin, W. Interferon-γ activates nuclear factor-κ B in oligodendrocytes through a process mediated by the unfolded protein response. PLoS ONE 2012, 7, e36408.
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866.
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227.
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575.
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life. Sci. 2022, 291, 120111.
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250.
- Lin, W.; Stone, S. Unfolded protein response in myelin disorders. Neural. Regen. Res. 2020, 15, 636–645.
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug. Discov. 2022, 21, 115–140.
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855.
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769.
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825.
- Lubetzki, C.; Zalc, B.; Williams, A.; Stadelmann, C.; Stankoff, B. Remyelination in multiple sclerosis: From basic science to clinical translation. Lancet Neurol. 2020, 19, 678–688.
This entry is offline, you can click here to edit this entry!