The function of the respiratory chain is closely associated with kidney function, and the dysfunction of the respiratory chain is a primary pathophysiological change in chronic kidney failure. The incidence of chronic kidney failure caused by defects in respiratory-chain-related genes has frequently been overlooked. Correcting abnormal metabolic reprogramming, rescuing the “toxic respiratory chain”, and targeting the clearance of mitochondrial reactive oxygen species are potential therapies for treating chronic kidney failure. These treatments have shown promising results in slowing fibrosis and inflammation progression and improving kidney function in various animal models of chronic kidney failure and patients with chronic kidney disease (CKD). The mitochondrial respiratory chain is a key target worthy of attention in the treatment of chronic kidney failure.
- mitochondrial respiratory chain
- chronic renal failure
- targeting
- therapeutic drugs
- energy substrate
- oxidative stress
1. Introduction
2. “Starting from Scratch”—Targeting the Energy Substrate Selection Stage
3. Strive for “Precision Strike”—Targeting the Mitochondrial Respiratory Chain
4. “Stepping on the Brake”—Targeting Mitochondrial Oxidative Stress
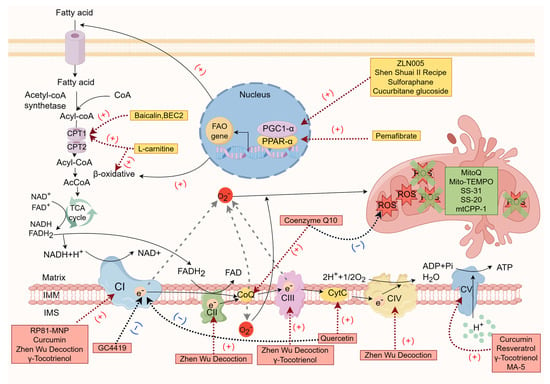
| Drug Name | The Main Action Stage | Mechanism | Current Usage Status |
|---|---|---|---|
| L-carnitine [15][16][17][18][19][20][21][22][23][24][93] | Energy substrate selection | Mediates fatty acid transport and promotes the tricarboxylic acid cycle | Validated by randomized clinical trials in patients with hemodialysis and peritoneal dialysis with chronic renal failure, but the results were controversial |
| ZLN005 [32][33] | Energy substrate selection | PGC-1α agonists, promotes fatty acid oxidation, mitochondrial biogenesis and function | Phenotypic improvement validation of mouse model of diabetes mellitus and UUO |
| Shen Shuai Ⅱ recipe [34] | Energy substrate selection | Activates PGC-1α and regulates mitochondrial dynamics | Phenotypic improvement validation of rat 5/6 nephrectomy CKD model |
| Sulforaphane [35] | Energy substrate selection | Enhances PGC-1α and NRF1 expression, improves lipid metabolism and mitochondrial biogenesis | Phenotypic improvement validation of rat UUO model |
| Cucurbitane glucoside [36] | Energy substrate selection | Activates PGC-1α | Lack of animal model validation |
| Pemafibrate [41] | Energy substrate selection | PPARα agonist, regulates fatty acid metabolism | Phenotypic improvement validation of mouse UUO and purine-induced CKD models |
| Baicalin, BEC2 [43][44] | Energy substrate selection | CPT1A agonist, accelerates β oxidation of long-chain fatty acids | Not verified by mouse CKD model |
| Coenzyme Q10 [47][48][49][50][51][52][53][54][55] | Mitochondrial respiratory chain | improves the electron transport efficiency of the respiratory chain, activates PGC-1α to improve fatty acid metabolism, and inhibits mitochondrial membrane potential depolarization | Phenotypic improvement validation of a rat renal hemirectomy CKD model and patients with chronic renal failure, large-scale clinical randomized controlled trials were lacking |
| RP81-MNP [56] | Mitochondrial respiratory chain | Upregulates the expression of mitochondrial complex I subunit and enhances the reduction state of complex I | Phenotypic improvement validation of cisplatin-induced mouse CKD model |
| GC4419 [57] | Mitochondrial respiratory chain | Inhibits mitochondrial complex I aberrant activity | Phenotypic improvement validation of cisplatin-induced mouse CKD model |
| MA-5 [58][59][60] | Mitochondrial respiratory chain | Promotes ATP synthase oligomerization and forms a supercomplex with mitofilin/Mic60 | Phenotypic improvement validation of cisplatin-induced mouse nephropathy model |
| Curcumin [63][64] | Mitochondrial respiratory chain | Maintains complexes I, V activity | Prophylactic administration was used to verify the protective effect of renal function in rat 5/6 nephrectomy CKD model |
| Quercetin [61][65][66] | Mitochondrial respiratory chain | Enhances cytochrome C concentration and inhibits the generation of superoxide anion by complex I | Validation of phenotypic improvement in rat UUO model |
| Resveratrol [70] | Mitochondrial respiratory chain | Increases the expression of ATP synthase β and cytochrome c oxidase subunit I protein, promotes ATP synthesis, and reverses mitochondrial hyperpolarization membrane potential | Validation of phenotypic improvement in rat 5/6 nephrectomy CKD model |
| ZhenWu Decoction [72] | Mitochondrial respiratory chain | Enhances mitochondrial respiratory complex I-V subunit expression to restore oxidative phosphorylation | Validation of phenotypic improvement in rat UUO model |
| γ-Tocotrienol [73] | Mitochondrial respiratory chain | Maintains complex I, III and F0F1-ATPase activity | Prophylactic administration has only been shown to be effective in a mouse model of ischemia–reperfusion acute kidney injury |
| MitoQ [78][79] | Mitochondrial oxidative stress | Targets mitochondria to prevent lipid peroxidation | Validation of phenotypic improvement in mouse aging model and chronic renal failure patients, large-scale clinical randomized controlled trials were lacking |
| Mito-TEMPO [80][81] | Mitochondrial oxidative stress | SOD enzyme mimics targeting ROS-mediated hypermethylation of the NDRG2 promoter | Validation of phenotypic improvement in mouse UUO model and rat 5/6 nephrectomy CKD model |
| SS-31 [86][87][88][89] | Mitochondrial oxidative stress | Targets the inner mitochondrial membrane to scavenge mitochondrial oxygen radicals by tyrosine or dimethyltyrosine residues | Validation of phenotypic improvement in rat 5/6 nephrectomy and UUO model |
| SS-20 [90] | Mitochondrial oxidative stress | Targets the inner mitochondrial membrane to scavenge mitochondrial oxygen radicals by tyrosine or dimethyltyrosine residues | Validation of phenotypic improvement in mouse 5/6 nephrectomy model |
| mtCPP-1 [92] | Mitochondrial oxidative stress | Targets mitochondria to scavenge mitochondrial oxygen radicals by dimethyltyrosine residues | Lack of animal model validation |
This entry is adapted from the peer-reviewed paper 10.3390/ijms25020949
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050.
- Eckardt, K.-U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Köttgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169.
- Huang, J.; Liang, Y.; Zhou, L. Natural products for kidney disease treatment: Focus on targeting mitochondrial dysfunction. Front. Pharmacol. 2023, 14, 1142001.
- Gaudry, S.; Verney, C.; Hajage, D.; Ricard, J.D.; Dreyfuss, D. Hypothesis: Early renal replacement therapy increases mortality in critically ill patients with acute on chronic renal failure. A post hoc analysis of the AKIKI trial. Intensive Care Med. 2018, 44, 1360–1361.
- Stoumpos, S.; Jardine, A.G.; Mark, P.B. Cardiovascular morbidity and mortality after kidney transplantation. Transpl. Int. 2015, 28, 10–21.
- Elshahat, S.; Cockwell, P.; Maxwell, A.P.; Griffin, M.; O’brien, T.; O’neill, C. The impact of chronic kidney disease on developed countries from a health economics perspective: A systematic scoping review. PLoS ONE 2020, 15, e0230512.
- O’Connor, P.M. Renal oxygen delivery: Matching delivery to metabolic demand. Clin. Exp. Pharmacol. Physiol. 2006, 33, 961–967.
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123.
- Mandel, L.J.; Balaban, R.S. Stoichiometry and coupling of active transport to oxidative metabolism in epithelial tissues. Am. J. Physiol. 1981, 240, F357–F371.
- Soltoff, S.P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 1986, 48, 9–31.
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646.
- Granata, S.; Gassa, A.D.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49.
- Popkov, V.A.; Silachev, D.N.; Zalevsky, A.O.; Zorov, D.B.; Plotnikov, E.Y. Mitochondria as a Source and a Target for Uremic Toxins. Int. J. Mol. Sci. 2019, 20, 3094.
- Li, S.-Y.; Susztak, K. The Role of Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC-1α) in Kidney Disease. Semin. Nephrol. 2018, 38, 121–126.
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717.
- Gülçin, I. Antioxidant and antiradical activities of L-carnitine. Life Sci. 2006, 78, 803–811.
- Marcovina, S.M.; Sirtori, C.; Peracino, A.; Gheorghiade, M.; Borum, P.; Remuzzi, G.; Ardehali, H. Translating the basic knowledge of mitochondrial functions to metabolic therapy: Role of L-carnitine. Transl. Res. J. Lab. Clin. Med. 2013, 161, 73–84.
- Hatanaka, Y.; Higuchi, T.; Akiya, Y.; Horikami, T.; Tei, R.; Furukawa, T.; Takashima, H.; Tomita, H.; Abe, M. Prevalence of Carnitine Deficiency and Decreased Carnitine Levels in Patients on Hemodialysis. Blood Purif. 2019, 47 (Suppl. S2), 38–44.
- Morgans, H.A.; Chadha, V.; Warady, B.A. The role of carnitine in maintenance dialysis therapy. Pediatr. Nephrol. 2021, 36, 2545–2551.
- Nishioka, N.; Luo, Y.; Taniguchi, T.; Ohnishi, T.; Kimachi, M.; Ng, R.C.; Watanabe, N. Carnitine supplements for people with chronic kidney disease requiring dialysis. Cochrane Database Syst. Rev. 2022, 12, CD013601.
- Maruyama, T.; Maruyama, N.; Higuchi, T.; Nagura, C.; Takashima, H.; Kitai, M.; Utsunomiya, K.; Tei, R.; Furukawa, T.; Yamazaki, T.; et al. Efficacy of L-carnitine supplementation for improving lean body mass and physical function in patients on hemodialysis: A randomized controlled trial. Eur. J. Clin. Nutr. 2019, 73, 293–301.
- Hamedi-Kalajahi, F.; Imani, H.; Mojtahedi, S.; Shabbidar, S. Effect of L-Carnitine Supplementation on Inflammatory Markers and Serum Glucose in Hemodialysis Children: A Randomized, Placebo-Controlled Clinical Trial. J. Ren. Nutr. Off. J. Counc. Ren. Nutr. Natl. Kidney Found. 2022, 32, 144–151.
- Fukuda, S.; Koyama, H.; Kondo, K.; Fujii, H.; Hirayama, Y.; Tabata, T.; Okamura, M.; Yamakawa, T.; Okada, S.; Hirata, S.; et al. Effects of nutritional supplementation on fatigue, and autonomic and immune dysfunction in patients with end-stage renal disease: A randomized, double-blind, placebo-controlled, multicenter trial. PLoS ONE 2015, 10, e0119578.
- Hamedi-Kalajahi, F.; Zarezadeh, M.; Mojtahedi, S.Y.; Shabbidar, S.; Fahimi, D.; Imani, H. Effect of L-carnitine supplementation on lipid profile and apolipoproteins in children on hemodialysis: A randomized placebo-controlled clinical trial. Pediatr. Nephrol. 2021, 36, 3741–3747.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012.
- Elsayed, E.T.; Nassra, R.A.; Naga, Y.S. Peroxisome proliferator-activated receptor-γ-coactivator 1α (PGC-1α) gene expression in chronic kidney disease patients on hemodialysis: Relation to hemodialysis-related cardiovascular morbidity and mortality. Int. Urol. Nephrol. 2017, 49, 1835–1844.
- Tamaki, M.; Hagiwara, A.; Miyashita, K.; Wakino, S.; Inoue, H.; Fujii, K.; Fujii, C.; Sato, M.; Mitsuishi, M.; Muraki, A.; et al. Improvement of Physical Decline Through Combined Effects of Muscle Enhancement and Mitochondrial Activation by a Gastric Hormone Ghrelin in Male 5/6Nx CKD Model Mice. Endocrinology 2015, 156, 3638–3648.
- Su, Z.; Klein, J.D.; Du, J.; Franch, H.A.; Zhang, L.; Hassounah, F.; Hudson, M.B.; Wang, X.H. Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am. J. Physiol. Ren. Physiol. 2017, 312, F1128–F1140.
- Feng, H.; Wang, J.-Y.; Yu, B.; Cong, X.; Zhang, W.-G.; Li, L.; Liu, L.-M.; Zhou, Y.; Zhang, C.-L.; Gu, P.-L.; et al. Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α Inhibits Vascular Calcification Through Sirtuin 3-Mediated Reduction of Mitochondrial Oxidative Stress. Antioxid. Redox Signal. 2019, 31, 75–91.
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347.
- Zhu, P.; Ma, H.; Cui, S.; Zhou, X.; Xu, W.; Yu, J.; Li, J. ZLN005 Alleviates In Vivo and In Vitro Renal Fibrosis via PGC-1α-Mediated Mitochondrial Homeostasis. Pharmaceuticals 2022, 15, 434.
- Zhang, L.-N.; Zhou, H.-Y.; Fu, Y.-Y.; Li, Y.-Y.; Wu, F.; Gu, M.; Wu, L.-Y.; Xia, C.-M.; Dong, T.-C.; Li, J.-Y.; et al. Novel small-molecule PGC-1α transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 2013, 62, 1297–1307.
- Wang, M.; Wang, L.; Zhou, Y.; Feng, X.; Ye, C.; Wang, C. Shen Shuai Ⅱ Recipe attenuates renal fibrosis in chronic kidney disease by improving hypoxia-induced the imbalance of mitochondrial dynamics via PGC-1α activation. Phytomed. Int. J. Phytother. Phytopharm. 2022, 98, 153947.
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; García-Arroyo, F.E.; Amador-Martínez, I.; Orozco-Ibarra, M.; Fernández-Valverde, F.; Pedraza-Chaverri, J. Sulforaphane Protects against Unilateral Ureteral Obstruction-Induced Renal Damage in Rats by Alleviating Mitochondrial and Lipid Metabolism Impairment. Antioxidants 2022, 11, 1854.
- Niu, B.; Ke, C.Q.; Li, B.H.; Li, Y.; Yi, Y.; Luo, Y.; Shuai, L.; Yao, S.; Lin, L.G.; Li, J.; et al. Cucurbitane Glucosides from the Crude Extract of Siraitia grosvenorii with Moderate Effects on PGC-1α Promoter Activity. J. Nat. Prod. 2017, 80, 1428–1435.
- Cheng, C.-F.; Chen, H.-H.; Lin, H. Role of PPARα and Its Agonist in Renal Diseases. PPAR Res. 2010, 2010, 345098.
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46.
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARα and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. JASN 2018, 29, 1223–1237.
- Jao, T.-M.; Nangaku, M.; Wu, C.-H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589.
- Horinouchi, Y.; Murashima, Y.; Yamada, Y.; Yoshioka, S.; Fukushima, K.; Kure, T.; Sasaki, N.; Imanishi, M.; Fujino, H.; Tsuchiya, K.; et al. Pemafibrate inhibited renal dysfunction and fibrosis in a mouse model of adenine-induced chronic kidney disease. Life Sci. 2023, 321, 121590.
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695.
- Zhang, M.; Xin, X.; Zhao, G.; Zou, Y.; Li, X.-F. In vitro absorption and lipid-lowering activity of baicalin esters synthesized by whole-cell catalyzed esterification. Bioorg. Chem. 2022, 120, 105628.
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905.
- Tang, C.; Dong, Z. Mitochondria in Kidney Injury: When the Power Plant Fails. J. Am. Soc. Nephrol. JASN 2016, 27, 1869–1872.
- Zhao, S.; Wu, W.; Liao, J.; Zhang, X.; Shen, M.; Li, X.; Lin, Q.; Cao, C. Molecular mechanisms underlying the renal protective effects of coenzyme Q10 in acute kidney injury. Cell. Mol. Biol. Lett. 2022, 27, 57.
- Yeung, C.K.; Billings, F.T.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 183.
- Ahmadi, A.; Begue, G.; Valencia, A.P.; Norman, J.E.; Lidgard, B.; Bennett, B.J.; Van Doren, M.P.; Marcinek, D.J.; Fan, S.; Prince, D.K.; et al. Randomized crossover clinical trial of coenzyme Q10 and nicotinamide riboside in chronic kidney disease. JCI Insight 2023, 8, e167274.
- Xu, Z.; Huo, J.; Ding, X.; Yang, M.; Li, L.; Dai, J.; Hosoe, K.; Kubo, H.; Mori, M.; Higuchi, K.; et al. Coenzyme Q10 Improves Lipid Metabolism and Ameliorates Obesity by Regulating CaMKII-Mediated PDE4 Inhibition. Sci. Rep. 2017, 7, 8253.
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.-Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal. 2014, 20, 2606–2620.
- Bakhshayeshkaram, M.; Lankarani, K.B.; Mirhosseini, N.; Tabrizi, R.; Akbari, M.; Dabbaghmanesh, M.H.; Asemi, Z. The Effects of Coenzyme Q10 Supplementation on Metabolic Profiles of Patients with Chronic Kidney Disease: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Curr. Pharm. Des. 2018, 24, 3710–3723.
- Drovandi, S.; Lipska-Ziętkiewicz, B.S.; Ozaltin, F.; Emma, F.; Gulhan, B.; Boyer, O.; Trautmann, A.; Xu, H.; Shen, Q.; Rao, J.; et al. Oral Coenzyme Q10 supplementation leads to better preservation of kidney function in steroid-resistant nephrotic syndrome due to primary Coenzyme Q10 deficiency. Kidney Int. 2022, 102, 604–612.
- Atmaca, M.; Gulhan, B.; Korkmaz, E.; Inozu, M.; Soylemezoglu, O.; Candan, C.; Bayazıt, A.K.; Elmacı, A.M.; Parmaksiz, G.; Duzova, A.; et al. Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr. Nephrol. 2017, 32, 1369–1375.
- Ishikawa, A.; Kawarazaki, H.; Ando, K.; Fujita, M.; Fujita, T.; Homma, Y. Renal preservation effect of ubiquinol, the reduced form of coenzyme Q10. Clin. Exp. Nephrol. 2011, 15, 30–33.
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850.
- Guo, X.; Xu, L.; Velazquez, H.; Chen, T.-M.; Williams, R.M.; Heller, D.A.; Burtness, B.; Safirstein, R.; Desir, G.V. Kidney-Targeted Renalase Agonist Prevents Cisplatin-Induced Chronic Kidney Disease by Inhibiting Regulated Necrosis and Inflammation. J. Am. Soc. Nephrol. JASN 2022, 33, 342–356.
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106.
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS ONE 2020, 15, e0231064.
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Matsuhashi, T.; Matsuo, A.; Sato, T.; Oba, Y.; Watanabe, S.; Minaki, D.; Saigusa, D.; et al. Mitochonic Acid 5 (MA-5), a Derivative of the Plant Hormone Indole-3-Acetic Acid, Improves Survival of Fibroblasts from Patients with Mitochondrial Diseases. Tohoku J. Exp. Med. 2015, 236, 225–232.
- Matsuhashi, T.; Sato, T.; Kanno, S.-I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic Acid 5 (MA-5) Facilitates ATP Synthase Oligomerization and Cell Survival in Various Mitochondrial Diseases. EBioMedicine 2017, 20, 27–38.
- Lagoa, R.; Graziani, I.; Lopez-Sanchez, C.; Garcia-Martinez, V.; Gutierrez-Merino, C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim. Biophys. Acta 2011, 1807, 1562–1572.
- Wang, D.; Yang, Y.; Zou, X.; Zheng, Z.; Zhang, J. Curcumin ameliorates CKD-induced mitochondrial dysfunction and oxidative stress through inhibiting GSK-3β activity. J. Nutr. Biochem. 2020, 83, 108404.
- Tapia, E.; Sanchez-Lozada, L.-G.; García-Niño, W.R.; García, F.E.; Cerecedo, A.; García-Arroyo, F.E.; Osorio, H.; Arellano, A.; Cristobal-Garcia, M.; Loredo, M.L.; et al. Curcumin prevents maleate-induced nephrotoxicity: Relation to hemodynamic alterations, oxidative stress, mitochondrial oxygen consumption and activity of respiratory complex I. Free Radic. Res. 2014, 48, 1342–1354.
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin prevents mitochondrial dynamics disturbances in early 5/6 nephrectomy: Relation to oxidative stress and mitochondrial bioenergetics. BioFactors 2017, 43, 293–310.
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1071–R1077.
- Jones, E.A.; Shahed, A.; Shoskes, D.A. Modulation of apoptotic and inflammatory genes by bioflavonoids and angiotensin II inhibition in ureteral obstruction. Urology 2000, 56, 346–351.
- Shoskes, D.; Lapierre, C.; Cruz-Corerra, M.; Muruve, N.; Rosario, R.; Fromkin, B.; Braun, M.; Copley, J. Beneficial effects of the bioflavonoids curcumin and quercetin on early function in cadaveric renal transplantation: A randomized placebo controlled trial. Transplantation 2005, 80, 1556–1559.
- Do Amaral, C.L.; Francescato, H.D.C.; Coimbra, T.M.; Costa, R.S.; Darin, J.D.C.; Antunes, L.M.G.; De Lourdes Pires Bianchi, M. Resveratrol attenuates cisplatin-induced nephrotoxicity in rats. Arch. Toxicol. 2008, 82, 363–370.
- Holthoff, J.H.; Wang, Z.; Seely, K.A.; Gokden, N.; Mayeux, P.R. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012, 81, 370–378.
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol improves mitochondrial function in the remnant kidney from 5/6 nephrectomized rats. Acta Histochem. 2017, 119, 392–399.
- Summerlin, N.; Soo, E.; Thakur, S.; Qu, Z.; Jambhrunkar, S.; Popat, A. Resveratrol nanoformulations: Challenges and opportunities. Int. J. Pharm. 2015, 479, 282–290.
- Zheng, M.; Hu, Z.; Wang, Y.; Wang, C.; Zhong, C.; Cui, W.; You, J.; Gao, B.; Sun, X.; La, L. Zhen Wu decoction represses renal fibrosis by invigorating tubular NRF2 and TFAM to fuel mitochondrial bioenergetics. Phytomed. Int. J. Phytother. Phytopharm. 2023, 108, 154495.
- Nowak, G.; Megyesi, J. γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia. Int. J. Mol. Sci. 2021, 22, 12674.
- Piko, N.; Bevc, S.; Hojs, R.; Ekart, R. The Role of Oxidative Stress in Kidney Injury. Antioxidants 2023, 12, 1772.
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384.
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412.
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596.
- Kirkman, D.L.; Stock, J.M.; Shenouda, N.; Bohmke, N.J.; Kim, Y.; Kidd, J.; Townsend, R.R.; Edwards, D.G. Effects of a mitochondrial-targeted ubiquinol on vascular function and exercise capacity in chronic kidney disease: A randomized controlled pilot study. Am. J. Physiol. Ren. Physiol. 2023, 325, F448–F456.
- Miao, J.; Liu, J.; Niu, J.; Zhang, Y.; Shen, W.; Luo, C.; Liu, Y.; Li, C.; Li, H.; Yang, P.; et al. Wnt/β-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell 2019, 18, e13004.
- Zhao, Y.; Fan, X.; Wang, Q.; Zhen, J.; Li, X.; Zhou, P.; Lang, Y.; Sheng, Q.; Zhang, T.; Huang, T.; et al. ROS promote hyper-methylation of NDRG2 promoters in a DNMTS-dependent manner: Contributes to the progression of renal fibrosis. Redox Biol. 2023, 62, 102674.
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free Radic. Res. 2017, 51, 237–252.
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253.
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Ortiz, A.; Sanchez-Niño, M.D. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13.
- Szeto, H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006, 8, E277–E283.
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690.
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1β and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. JASN 2017, 28, 1437–1449.
- Zhao, H.; Liu, Y.-J.; Liu, Z.-R.; Tang, D.-D.; Chen, X.-W.; Chen, Y.-H.; Zhou, R.-N.; Chen, S.-Q.; Niu, H.-X. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur. J. Pharmacol. 2017, 804, 57–67.
- Mizuguchi, Y.; Chen, J.; Seshan, S.V.; Poppas, D.P.; Szeto, H.H.; Felsen, D.; Hou, Y.; Li, S.; Wu, M.; Wei, J.; et al. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2008, 295, F1545–F1553.
- Liu, Z.-R.; Chen, S.-Q.; Zou, Y.-W.; Wu, X.-Y.; Li, H.-Y.; Wang, X.-Q.; Shi, Y.; Niu, H.-X. Hypochlorite modified albumins promote cell death in the tubule interstitium in rats via mitochondrial damage in obstructive nephropathy and the protective effects of antioxidant peptides. Free Radic. Res. 2018, 52, 616–628.
- Sun, L.; Xu, H.; Wang, Y.; Ma, X.; Xu, Y.; Sun, F. The mitochondrial-targeted peptide SBT-20 ameliorates inflammation and oxidative stress in chronic renal failure. Aging 2020, 12, 18238–18250.
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 2019, 211, 57–67.
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4589–4599.
- Sabry, M.M.; Ahmed, M.M.; Maksoud, O.M.A.; Rashed, L.; Morcos, M.A.; El-Maaty, A.A.; Galal, A.M.; Sharawy, N. Carnitine, apelin and resveratrol regulate mitochondrial quality control (QC) related proteins and ameliorate acute kidney injury: Role of hydrogen peroxide. Arch. Physiol. Biochem. 2022, 128, 1391–1400.