Cardiovascular diseases (CVD) refer to a range of disorders that specifically impact the circulating system (heart and vessel network). According to the World Health Organization (WHO), CVD is a major cause of death, accounting for an estimated 17.9 million people every year and representing approximately 30% of deaths worldwide (WHO 2023). Myocardial fibrosis represents a dynamic restructuring of the extracellular matrix (ECM) in response to cardiac stress. Two types of mechanisms have been involved in cardiac fibrosis: “replacement” or “reparative” fibrosis is implicated in the reparative mechanisms that unfold during the acute phase of injuries, such as myocardial infarctions, pulmonary arterial hypertension or congenital heart disease, in order to locally replace dying cardiomyocytes [
1]. In stark contrast, “reactive” or “diffuse” myocardial fibrosis corresponds to ECM remodeling developing gradually over time in response to chronic pressure overload and results in the progressive deposition of collagen in the interstitial and perivascular regions [
1,
2,
3]. This persistent collagen deposition disrupts the normal function of either the left or the right ventricle of the heart, leading to ventricular stiffness and poor electrical conductance of the myocardium. Left ventricle reactive fibrosis occurs more specifically in chronic pathological conditions, such as systemic hypertension, aortic stenosis, diabetes mellitus [
2], obesity [
4], and cardiorenal syndrome [
5] and is a key contributing factor leading to arrhythmias and progression to heart failure [
1]. Therefore, investigation of the cellular and molecular mechanisms involved in the initiation and progression of myocardial fibrosis is of major importance to prevent its onset in high-risk but not yet diseased patients.
2. Endothelial to Mesenchymal Transition in Cardiovascular Diseases
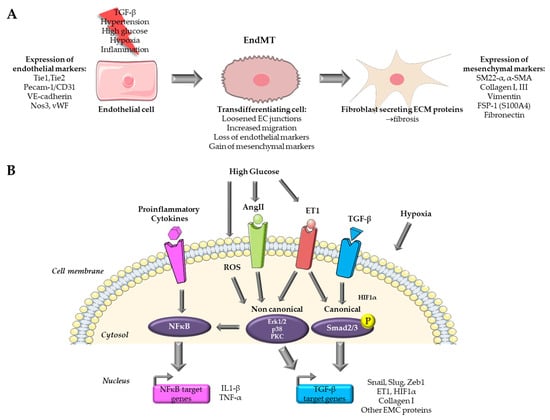
Endothelial-mesenchymal transition is a complex process that involves a transition from endothelial cells (ECs) towards a mesenchymal-like phenotype in response to a range of specific stimulators. Cells undergoing EndMT adopt a spindle-shaped morphology that facilitates their unrestricted movement within the ECM because of the loss of their cellular polarity and downregulation of adherent and tight junction proteins. This mobility enables them to assemble the connective tissue crucial for organ function [
8]. EndMT mainly occurs during embryonic development, particularly in the early stages of cardiac septum and valve formation [
8,
9]. It is generally accepted that this transition is characterized by the loss of endothelial markers, such as platelet endothelial cell adhesion molecule (Pecam-1/CD31), vascular endothelial cadherin (VE-cadherin), von Willebrand factor (vWF), and the acquirement of a mesenchymal (fibroblastic-like) phenotype, characterized by the expression of mesenchymal markers including SM22-α (Smooth muscle protein 22-alpha), vimentin, α-SMA (Actin alpha 1 skeletal muscle), type I collagen, FSP-1 (Fibroblast specific protein-1, i.e., S100A4), fibronectin [
6,
8] (
Figure 1A). Nevertheless, when examining EndMT at the molecular level, there is currently no consensus on criteria allowing the precise definition of this process. This lack of standardization poses a growing challenge, as it leads to a lack of uniformity and limited comparability of data issued from various model systems and research publications [
9].
Figure 1. Overview of pathways targeted for the study of EndMT in cardiovascular diseases. (A) Endothelial to mesenchymal transition process. (B) Pathways targeted in in vitro models for the study of EndMT. Parts of the figure were drawn by using pictures from Servier Medical Art.
Several signaling pathways have been documented to initiate and regulate EndMT during both physiological and pathological conditions, including bone morphogenetic protein (BMP)–transforming growth factor (TGF), vascular endothelial growth factor A (VEGFA), epidermal growth factor (EGF), fibroblast growth factor (FGF), Notch, platelet-derived growth factor (PDGF), Wnt/β-catenin, calcineurin–NFAT, transcription factor GATA4-mediated transcriptional regulation, endothelin-1 (ET-1) and inflammatory signaling. These pathways have been extensively studied in the latest reviews [
2,
6,
8,
9]. However, the implication of EndMT in fibrosis remains unclear.
3. Existing In Vitro Models of Endothelial to Mesenchymal Transition in Cardiovascular Diseases
The majority of in vitro models for EndMT utilize primary ECs. In vitro models offer several advantages over their in vivo counterparts, including ease of accessibility, cost-effectiveness, and the potential for achieving highly reproducible outcomes. Nevertheless, it is important to acknowledge that in vitro models do have limitations when compared to in vivo models, and these limitations may impact the extrapolation of findings derived from such models. An overview of the signaling pathways targeted by in vitro models for the study of EndMT in cardiovascular diseases is presented in Figure 1B.
3.1. Cytokine Based In Vitro Models
3.1.1. TGF-β Signaling
Among the growth factor families, TGF-β emerges as the most significant cytokine-inducing transdifferentiation of EC (recent review [
19]). TGF-β plays a pivotal role at the early embryonic stages in guiding the atrioventricular canal formation [
20]. TGF-β is secreted by both fibroblasts and macrophages and stimulates collagen and ECM component production by enhancing EndMT, which in turn leads to fibrotic lesions [
21]. The initiation of TGF-β-induced EndMT signalization following the canonical pathway is achieved through the binding of TGF-β isoforms to its receptor TGFBR1/2, activating its intrinsic kinase activity. This cascade of kinase/phosphorylation activity continues within the cell to the nucleus, across phosphorylation of downstream effectors, including receptor-activated Smad (R-Smads) and particularly Smad2/3, leading to the formation of a hetero-oligomeric complex with Smad4. This promotes their translocation in the nucleus where the complex binds to target gene promoters, inducing the expression of transcription factors like Snail, Slug, and Twist, which leads to the up-regulation of numerous EndMT-involved genes [
8,
19]. As for the canonical pathway, emerging evidence highlights the involvement of non-canonical pathways in the signaling of TGF-β-induced EndMT through the activation of a Smad-independent kinase complex, which requires several other downstream effectors, such as the mitogen-activated protein kinase (MAPK), PI3K, and PKC-δ. Indeed, TGF-β has been shown to induce the activation of MAPK cascades in a fibrosis context, triggering extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK. These activated downstream effectors can transduce signals to the nucleus, leading to an increased expression of transcription factors, thereby stimulating the up-regulation of EndMT-associated gene expression [
19]. While the precise mechanisms governing these pathways exhibit diversity and remain partially understood, the majority of these pathways ultimately intersect by modulating transcription factors, such as Snail and Twist, to inhibit the expression of EC markers and to induce the expression of mesenchymal proteins.
3.1.2. TGF-β Isoform Specific Effects to Induce EndMT
Varying degrees of potency in promoting EndMT has been attributed to the three TGF-β isoforms, namely TGF-β1, TGF-β2, and TGF-β3. Among them, TGF-β2 appears to be the isoform with the greatest potential for inducing EndMT, as highlighted during embryonic heart development [
22] and on immortalized human dermal microvascular endothelial cells (HMVEC) [
23,
24]. Exposure for 72 h to 1 ng/mL TGF-β1 and TGF-β3 led to an increase in the expression of TGF-β2, while TGF-β2 treatment did not. This suggests that EndMT induced by TGF-β1 and TGF-β3 is the result of secondary effects mediated through the secretion of TGF-β2. Moreover, TGF-β2 increases phosphorylation (activation) of Smad2/3 (canonical) and p38 MAPK (non-canonical) at a higher level than TGF-β1 and TGF-β3. Finally, silencing TGF-β2 attenuated the expression of EndMT markers in cells treated with TGF-β1 and TGF-β3. This, therefore, suggests that TGF-β1 and TGF-β3-induced EndMT involves a paracrine loop mediated by TGF-β2 [
23]. Interestingly, studies carried out over the last 10 years have used either TGF-β2 [
23,
24,
25,
26] or TGF-β1 on endothelial cells from different origins [
27,
28,
29,
30,
31,
32,
33,
34] and, more rarely, TGF-β3 [
23,
35].
3.1.3. EndMT Induction Level Depend on Endothelial Cell Origin
Most in vitro models use endothelial cells of different anatomical origins treated with a member of the TGF-β family. In these models, EndMT is characterized by four characteristics: (1) simultaneous expression of endothelial and mesenchymal markers, (2) heightened cellular migratory capabilities, (3) diminished expression of endothelial traits, including reduced leukocyte adhesion and impaired tubule formation, and (4) increased manifestation of mesenchymal/myofibroblastic traits, such as augmented collagen production and enhanced contractility [
36]. Several EC models are widely used, such as human umbilical vein ECs (HUVEC), aortic EC (HAEC), coronary EC (coronary artery CAEC, or microvascular CMVEC), cardiac EC (microvascular CMEC, atrial endocardial AEEC), pulmonary artery EC (PAEC), and dermal EC (HMEC). Two studies provide interesting evidence that cells from different anatomical origins do not have the same potential to enter transdifferentiation. Among the origins tested (coronary artery, aortic, umbilical vein, pulmonary artery), cells of aortic and coronary origin showed the best responses to induction by TGF-β2 treatment. This results in an increase in several mesenchymal markers, such as type I collagen, actin alpha cardiac muscle (ACTC), SM22-α, Calponin 1 (CNN1), Snail, and maintenance of endothelial markers such as Pecam-1 and VE-cadherin. This is also accompanied by a morphological and functional change, with a diminished ability to form vessel-like structures [
25]. In cells of aortic origin, the induction of EndMT is accompanied by significant activation of the non-canonical pathway via ERK1/2. In aortic and coronary cells, induction is potentiated by Snail overexpression in combination with TGF-β2 treatment, which is less obvious for HUVEC [
37] or PAEC [
25,
37,
38]. Moreover, cells of umbilical and pulmonary origin showed a delay or resistance to enter transdifferentiation and do not produce type I collagen (something also observed in the laboratory, unpublished data). It is, therefore, clear that the origin of EC plays a role in their ability to enter EndMT and that more information is required about these origins, as well as the conditions under which EndMT is induced.
3.1.4. Induction of EndMT by Co-Treatment with Proinflammatory Signals
The second most widely used model is that of EC treated with a combination of TGF-β and a proinflammatory cytokine, like IL1-β, TNF-α, or both. Indeed, the inflammatory context found in many cardiovascular diseases has been shown to promote cardiac fibrosis [
3]. Most studies demonstrate that induction of EndMT is possible using TGF-β isoforms in combination with a proinflammatory cytokine [
35,
38,
39]. This is the case for HUVEC, where costimulation with IL-1β and TGF-β1 or 2 caused synergistic induction of EndMT accompanied by overexpression of the transcription factor NF-κB, an increase in its nuclear translocation [
40,
41], and type I collagen expression, which was also the case in PAEC [
32,
38]. Cotreatment with TNF-α also induced EndMT [
42]. Treatment with IL-1β and TNF-α without TGF-β was also able to induce expression changes related to EndMT in HUVEC [
41,
43], suggesting that these cells may be more susceptible to inflammatory stimuli. This was also demonstrated in other organ fibrosis disorders, such as intestinal fibrosis [
44]. However, in that particular case, no significant change in their migratory capacity was observed, confirming a resistant or delayed capacity of entering EndMT in HUVEC and PAEC [
38], showing once again that cell origin plays a role in the ability to enter EndMT. Finally, angiotensin II (Ang II), a major fibrosis inducer [
3], is also able to induce EndMT by activating NFκB signaling and pro-inflammatory cytokine production in HUVEC [
45,
46,
47,
48] in HMVEC [
49] and in HCAEC [
50]. AngII also induces and activates the TGF-β axis [
3]. In short, proinflammatory signals, whether or not associated with TGF-β, are capable of inducing EndMT in ECs and represent widely used in vitro models.
3.2. Hypoxia-Based In Vitro Models
Chronic hypoxia plays a pivotal role in the development of cardiac fibrosis and is induced by a reduced density of microvessels, which subsequently impacts oxygen delivery and increased oxygen consumption due to the activation of inflammatory cells and fibroblasts. Chronic hypoxia independently contributes to abnormal ventricular remodeling and the onset of cardiac fibrosis [
51]. Hypoxia has the capability to trigger EndMT via HIF1 signaling, even in the absence of TGF-β, as it directly regulates Snail expression through HIF1α in HCAEC [
52]. Several publications, such as MVEC [
53], HCAEC [
52], and HUVEC [
54,
55,
56], use cells under hypoxia as an EndMT model. Interestingly, hypoxia is widely used on PAEC [
57,
58,
59] and PMVEC of pulmonary origin [
60]. Indeed, extensive vascular remodeling occurs in pulmonary arterial hypertension (PAH), where neointimal and medial thickening results from fibrosis in the pulmonary arteries [
12]. In vitro models of hypoxia-induced EndMT were developed in the last 10 years and allowed elucidation of signaling cascades activated in response to hypoxia in PAH (reviewed in [
12]). Hypoxia conditioning of EC, therefore, appears to be a relevant in vitro model for studying EndMT, especially in PAH.
3.3. High Glucose Based In Vitro Models of Organ Fibrosis in Diabetes Mellitus
One of the consequences of hyperglycemia is vascular dysfunction, which is a main component of diabetes mellitus, leading to both micro- and macro-vascular complications, and which affects several organs such as the kidney (diabetic nephropathy), heart (diabetic cardiomyopathy), retina (diabetic retinopathy), resulting in organ fibrosis. EndMT has been identified as an early important potential trigger in diabetic complications, as ECs are among the first to be damaged by hyperglycemia (recent review in [
17]). High glucose activates a variety of EndMT-inducing pathways, such as TGF-β (through the PKC pathway), ET-1, and inflammatory signals. EndMT models were therefore designed with EC from these different origins and treated with high glucose concentrations in order to mimic hyperglycemia. HUVEC is widely used as a model for diabetes-associated cardiac fibrosis [
14,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70]. In particular, Yu et al. showed that 24 h treatment with 30 mM glucose resulted in the expression of the mesenchymal markers α-SMA and type I collagen in HUVEC. Endothelial markers (Pecam-1, VE-cadherin) were under-expressed more slowly or at higher glucose concentrations, reflecting a delay in the regulation of these markers during glucose-mediated EndMT. Finally, mesenchymal cells were obtained at 60 mM glucose and after 48 h of treatment. High glucose-induced TGF-β1 production from 48 h of treatment [
63] shows that high glucose-mediated EndMT involves the TGF-β pathway. In vitro studies using HUVEC are usually combined with in vivo studies on animal models of diabetic disease. Aortic EC and CAEC are also used to study diabetic cardiomyopathy [
71,
72,
73,
74,
75]. It is worth noting that to study EndMT in diabetic nephropathy and kidney fibrosis, glomerular endothelial cells (GEnC) are specifically used [
76,
77,
78,
79,
80,
81,
82,
83]. Finally, human retinal EC is used to study diabetic retinopathy [
84,
85,
86,
87,
88].
Recently, a new 3D model of the aortic valve was developed in order to enhance the comprehension of the dynamic interplay between ECs and their microenvironmental matrix in the context of hyperglycemia [
89]. The authors used hydrogel containing extracellular matrix from porcine aortic root and human valve cells. They cultivated valve EC on the hydrogel’s surface and valve interstitial cells within the hydrogel, then exposed this 3D structure to high glucose conditions. The authors observed a reduced expression of endothelial markers Pecam-1 and VE-cadherin and increased expression of mesenchymal markers α-SMA and Vimentin. There was also a loss of intercellular junctions and an enhanced expression of inflammatory molecules. Finally, valvular ECs showed enhanced monocyte adhesion via a mechanism involving adhesion molecules such as ICAM-1 and VCAM-1, showing dysfunctionality. The 3D model contains an ECM mainly composed of collagen I and III, similar to the aortic valve structure [
89]. This model could also be interesting for cardiac myocardial fibrosis because ECM has a similar composition.