Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Fabry Disease (FD) is a rare lysosomal storage disorder caused by mutations in the GLA gene on the X chromosome, leading to a deficiency in α-galactosidase A (AGAL) enzyme activity. This leads to the accumulation of glycosphingolipids, primarily globotriaosylceramide (Gb3), in vital organs such as the kidneys, heart, and nervous system.
- Fabry disease
- GLA gene
- genetic basis
- X-chromosome inactivation
1. Introduction
Fabry disease is a multisystemic, lysosomal storage disease caused by mutations of the GLA gene mapping on chromosome X [1,2]. GLA is a 14-kb gene located on Xq22.1 that encodes the α-galactosidase A (AGAL) enzyme [3]. It is a polypeptide of 429 amino acids, and the Human Gene Mutation Database lists over 900 variants, of which 69% account for missense mutations [4]. Deficiency in the AGAL enzyme leads to the degradation of glycosphingolipids from cell membranes and causes the accumulation of globotriaosylceramide (Gb3) and other lipid complexes, mostly in vital organs (kidneys, heart, and nervous system) [2]. FD was first described by Johannes Fabry and William Anderson in 1898 [5,6].
FD was initially thought to predominantly affect males, having a single uncompensated mutated GLA gene. This could result in a severe lack of AGAL activity and lead to the early emergence of symptoms during childhood/adolescence with progressive organ damage and increased death risk [7]. Due to the X-linked inheritance, heterozygous females were considered to be asymptomatic carriers; however, over time, it has become evident that females exhibit a diverse range of clinical symptoms and experience a level of clinical severity often equivalent to that observed in males [8,9]. The reasons behind the diversity in the female phenotype are not yet well understood, although the potential influence of X-chromosome inactivation (XCI) has been postulated. The variation in the phenotype is a result of a process known as “lyonization”, in which one of the two X chromosomes in somatic cells is randomly inactivated [2].
2. FD Inheritance in Females
Initially, FD was considered to be an X-linked recessive hereditary disorder that affected primarily hemizygous males [22], whereas females were thought to be the carriers who were either asymptomatic or to have mild symptoms [23]. However, research over the years has shown that most females carrying the mutated gene may exhibit signs and symptoms of the disease [22]. It was observed that heterozygous females could be affected similarly to hemizygous males, and thus FD was postulated to be classified as X-linked [24]. Consequently, FD is now referred to as an “X-linked transmission” disease, and the use of the term “carrier” for women carrying a disease-causing mutation may no longer be correct [25]. The GLA gene mutations are typically heterozygous in females. Although a female can have the defective gene on both X chromosomes, the homozygous form is incredibly uncommon [26]. Regardless of sex, an affected woman who is heterozygous for the GLA gene mutation has a 50% probability of transferring a faulty gene to both males and females (27). (See Figure 1).
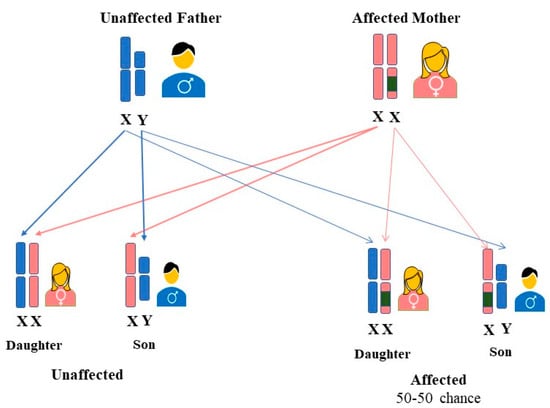
Figure 1. Inheritance pattern of FD in women. A mother with a GLA variant on one of her two X chromosomes has a 50% probability of passing FD to each of her offspring, regardless of their gender.
The random inactivation of one of the two X chromosomes, also known as lyonization, may have an impact on women just as severely as it does on hemizygous men [4]. The activity of AGAL in afflicted women can range from low to normal as a result of lyonization [3]. It has been proposed that XCI may have a potential role in the phenotypic expression of FD in females. In heterozygous females, the disease’s phenotypic diversity is substantially higher than in males, and symptoms typically appear late in life and progress gradually [24]. The rate of disease progression differed among females with the same pathogenic variants, perhaps because of the degree of skewed XCI that favors the mutant GLA allele [27]. Studies investigating the prevalence of skewed XCI in females with FD have been published; however, they are ambiguous and contradictory.
Genotype-phenotype correlations in Fabry disease have been a subject of increasing interest in the scientific community. The Lyon hypothesis, or random X-inactivation, has been proposed as a potential explanation for the phenotypic expression of Fabry disease in heterozygous women [28]. The wide heterogeneity in the natural history of patients with Fabry disease has led to efforts to establish potential associations between genotypes and phenotypic expressions of GLA gene [29]. However, reports have indicated that the same mutation can cause different phenotypes in the same family and different mutations can lead to the same phenotype, implying that no clear genotype-phenotype correlation could be identified in Fabry disease [30].
Female heterozygous patients with FD had varying degrees of disease severity, and it became worse as they aged [31]. The phenotypic expression of FD in females is significantly influenced by the degree of XCI [32]. Some investigations identified a correlation between the severity of disease manifestation and the pattern of XCI [33,34,35]. However, others do not corroborate these observations [36,37]. So many authors have dismissed its significance in elucidating and explaining phenotypic variability. The findings of previously published research indicate a correlation between clinical index and X inactivation, with two of four heterozygotes carriers of FD presenting a skewed XCI in favor of the mutant allele and the other two asymptomatic females exhibiting a skewed XCI favoring the wild-type allele [33]. On the contrary, a different study found no variations in the XCI ratio between 28 FD-affected women and healthy women of the same age. Skewed X inactivation was present in only 18% of heterozygous females [36]. Elstein et al. observed similar results among 77 heterozygous females, where only 18.2% had a significantly skewed XCI [37]. These findings do not provide significant evidence for the notion that skewed XCI is related to the severity of disease manifestation among female carriers of the mutant GLA gene.
Classical methods for measuring X-chromosome inactivation (XCI) skewness may not be sufficient to fully explain the manifestations of FD in women. In addition to unbalanced XCI, allele-specific DNA methylation at the promoter of the GLA gene can influence the expression levels of the mutated allele, thereby effecting the onset and outcome of FD. DNA methylation is an epigenetic modification that occurs at both X and autosomal genes, primarily at cytosine residues of CpG sites, and can regulate gene expression [38]. Therefore, analyzing DNA methylation at the GLA promoter, specifically distinguishing between the mutated and non-mutated alleles, can provide more informative insights. Recent studies have shown that DNA methylation can help in explaining the variability in the clinical phenotype observed in FD [39]. A study by Hossain et al., [40] investigated the linkage between DNA methylation patterns and Fabry disease severity in 36 affected women. They analyzed DNA methylation in peripheral blood and skin fibroblasts using methylation-sensitive restriction enzymes and bisulfite Sanger sequencing. The research identified a distinct correlation among the severity of the phenotype, the accumulation of lysoGb3, and methylation of the normal allele detected by non-digestion with methylation-sensitive restriction enzymes [39].
In a separate investigation, Hossain et al., [41] explored the impact of DNA methylation on the Fabry phenotype. The researcher underscored the significance of unbalanced DNA methylation in the expression of mutated alleles for the GLA gene, which can exert a substantial impact on the clinical phenotype. This was demonstrated in a severe case of a heterozygous female Fabry patient who manifested various symptoms, including acroparesthesia, facial dysmorphism, left ventricular hypertrophy, and intellectual disability.
3. FD Symptoms in Women
FD is a multisystem disorder that starts with cellular dysfunction, progresses over several years, and eventually causes organ functional impairment [100]. The classic type of FD manifests in childhood or adolescence and is characterized by angiokeratomas and corneal degeneration caused by endothelial dysfunction [101,102]. There is a substantial intra- and interfamilial variance in age of onset, clinical features, and clinical course. The typical clinical manifestation of FD in males has received extensive research [2]. In the past, it was believed that FD mostly affected men, while women were thought to be asymptomatic carriers. However, over the past decade, there has been a significant shift in the understanding of FD manifestation in females. Recent studies have conclusively demonstrated that the condition is not restricted to male manifestations only [103,104]. Recent studies have recognized that a significant proportion of female FD heterozygotes experience complications, albeit typically in an attenuated form compared to male FD [105]. The presentation of the disease in women varies widely, with clinical phenotypes ranging from asymptomatic individuals to women presenting severity comparable to classically affected men. This variability extends not only to the intensity of symptoms but also to the range of affected tissues and the onset of symptoms. Notably, the onset of initial symptoms and complications in adulthood tends to occur later in females compared to males [11].
The main initial clinical manifestations in female heterozygous FD individuals encompass renal, cardiac, and cerebrovascular symptoms, along with gastroenterological disturbances, corneal opacities, and angiokeratoma [104,105,106]. Other symptoms in female FD patients depend on the involvement of the gastrointestinal system, often abdominal pain, diarrhea, or constipation [107] (See Figure 2). Apart from pain, exhaustion, exercise intolerance, and a decreased intake of oxygen are frequently experienced by heterozygous Fabry women [11]. FD appears to be an interesting genetically determined model of chronic kidney disease, chronic neurodegenerative disease, and chronic heart failure (CHF) to investigate the mechanisms of progression of these common diseases [108,109]. Apart from other clinical manifestations, a retrospective analysis of pregnancies among women with FD revealed an increased prevalence of specific pregnancy complications when compared to the broader population of pregnant women. In a study led by Holmes et al., Fabry-affected women completed a survey, revealing that certain Fabry-related symptoms and characteristics, such as gastrointestinal problems, acroparesthesias, proteinuria, headaches, and postpartum depression, could exacerbate during pregnancy. However, there were no documented life-threatening consequences associated with these complications [110]. Specific concerns related to females with FD during pregnancy involve the potential impact of microvascular disease, which may elevate the risk of clotting and exacerbate renal function [9,27]. Also, Gb3 storage has been noted in both maternal and fetal placental tissues, heightening the susceptibility to constrictions in placental blood vessels [111,112,113]. Additionally, there are apprehensions regarding the condition of an affected fetus, leading to pregnancy complications attributed to abnormal Gb3 storage [113]. Concomitant conditions, such as preeclampsia, gestational diabetes, hypertension, and maternal age at delivery, may further complicate pregnancy in women with pre-existing FD [114]. In line with the prevailing Fabry disease guidelines, the survey reinforces the suggestion that pregnant women with FD should undergo assessment and, in certain instances, be monitored during pregnancy by a maternal-fetal specialist, in addition to receiving standard prenatal care [110]. However, further research is still needed to fully understand the specific effects and implications of FD on pregnancy health and outcomes for affected women.
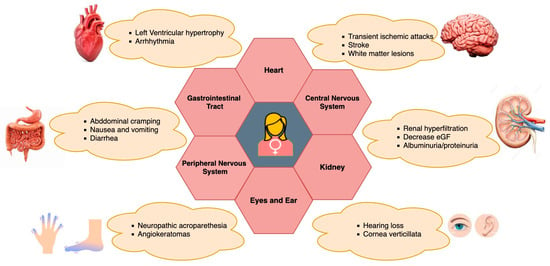
Figure 2. Clinical manifestations of FD in female heterozygotes and its multiorgan involvement.
Regarding infertility, a study by Laney et al. [115] revealed that among 197 females with Fabry disease (FD), 40 encountered infertility issues, and 31 out of 225 females sought an infertility assessment to address specific concerns related to infertility. The prevalence of infertility in females with FD surpasses the general population rate, where 12% of women aged 25–44 reported ever encountering infertility issues. However, it is comparable to the number of females in the general population undergoing infertility evaluations [116,117,118]. Out of the 242 females with FD, 30 (12.4%) utilized infertility services in attempts to conceive, employing methods such as donor eggs, in vitro fertilization, and hormonal treatments to enhance ovulation. It is noteworthy that some females opted for Pre-implantation Genetic Screening (PGS) for FD and donor eggs, not due to infertility but to prevent the transmission of FD [115]. Regarding menstrual pain, there is limited specific research and no direct evidence to suggest that menstrual pain is increased in FD women. In a study by Bouwman et al. [119], no substantial difference in the prevalence of premenstrual symptoms, menstrual symptoms, or menarche was observed between females with Fabry disease and the control group. Nevertheless, a higher incidence of reported loss of libido was noted among Fabry disease females compared to the controls. It is important to note that loss of libido may be influenced by the presence of a chronic medical condition [120] and may not necessarily be Fabry-specific. In conclusion, Fabry disease can have implications for pregnancy, infertility, and potentially menstrual health in affected women. However, further research is needed to fully understand the specific effects and implications of FD on these aspects of women’s health.
Renal impairment is one of the prominent features of FD, in which initial indexes include impaired GFR, proteinuria, and tubular derangements [121]. In male hemizygous patients, renal failure is a prominent cause of premature death [122]. In comparison, only 1–2% of females require dialysis or a kidney transplant due to the significantly lower incidence of renal failure in females that leads to end-stage renal disease (ERSD) [28]. A case report of a 26-year-old female patient with FD was published by Kriegsmann and colleagues, where histological, immunohistochemical, and electron microscopic analysis of a kidney biopsy revealed glomerulonephritis, extracapillary proliferative (crescentic), and granulomatous interstitial nephritis [123]. Fabry nephropathy in females is more common and varied than previously believed, according to a thorough review of renal data from 1262 untreated patients (54% of whom were female) included in the Fabry Registry [106]. At the same mean age as males, a considerable number of females had developed moderate to severe renal dysfunction [124]. The median age of occurrence for females with renal progression has been observed to be 38 years [125].
In accordance with MacDermot et al. [105] and Whybra et al. [126], women with FD frequently exhibit cardiac involvement and evidence of structural cardiac damage, especially left ventricular hypertrophy (LVH) cardiomyopathy and mitral valve insufficiency. This constitutes an important contributor to morbidity and mortality [42]. The majority of female Fabry cardiomyopathy patients possess additional characteristics, such as replacement fibrosis and compromised regional myocardial function [127]. Globotriaosylceramide accumulates in the heart when there is cardiac involvement in FD-affected women, leading to cardiac hypertrophy that could deteriorate and result in diastolic filling impairment and contractility impairment. Atrioventricular conduction abnormalities, arrhythmias, coronary insufficiency, and valvular involvement are other potential symptoms of cardiac involvement [42]. Cardiovascular events manifested at a median age of 47 years, which is higher than the average age of men (41 years) [124]. A range of symptoms, such as peripheral neuropathy, pain, autonomic dysfunction, and central nervous system manifestations, have been associated with neurological involvement in FD-affected women. As the disease progresses, both the peripheral and autonomic nerve systems are affected, resulting in paresthesia and neuropathic pain [8]. In females with FD, stroke and transient ischemic attacks (TIAs) are quite prevalent, and they are likely to be brought on by a combination of endothelial dysfunction and aberrant vascular function regulation [27]. The initial screening study for adolescents with cryptogenic stroke was published in 2005 and indicated a significant number of patients comprising a diagnosis, with 2.4% being females [127]. In a published study, it was found that female patients developed cerebrovascular complications from stroke and TIAs more frequently than male patients (27% compared with 12%) [28]. Such problems have been reported in 5–27% of heterozygous females [8,9,10]. About one-third of female patients with neurovascular conditions reported additional symptoms such as vertigo, tinnitus, hearing loss, and hyperhidrosis. Galanos et al. hypothesized that anhidrosis in females may be an indication of later serious renal disease [128]. The cerebrovascular symptoms in females appear to start at an average age of 43 years, thus later than in males (age, 38 years) [124].
This entry is adapted from the peer-reviewed paper 10.3390/genes15010037
This entry is offline, you can click here to edit this entry!