Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Parkinson’s disease (PD) is the second most prevalent neurodegenerative movement disorder worldwide, which is primarily characterized by motor impairments. Even though multiple hypotheses have been proposed over the decades that explain the pathogenesis of PD, presently, there are no cures or promising preventive therapies for PD. This could be attributed to the intricate pathophysiology of PD and the poorly understood molecular mechanism. To address these challenges comprehensively, a thorough disease model is imperative for a nuanced understanding of PD’s underlying pathogenic mechanisms.
- Parkinson’s disease
- α-synuclein
- molecular mechanism
- genetics of Parkinson’s disease
1. Introduction
Despite the fact that cases of PD are typically sporadic, various mutations are linked to the disease pathogenesis. They represent roughly 2–3% of late-onset forms of PD and more than 50% of patients with early onset. A clearer understanding of the molecular etiology of hereditary PD has been made possible by discovering gene mutations connected to the onset of familial cases of PD. A long list of genes is known to contribute to PD, and many more may yet be discovered. Mainly, six genes have been explored and linked to heritable PD. Whilst Parkin, DJ-1, PINK1, and ATP13A2 mutations are passed down in an autonomously recessive manner, SNCA and Leucine-rich repeat kinase 2 (LRRK2) mutations are passed down in an autosomal dominant form [3]. General gene-based molecular mechanisms in the pathogenesis of PD have been illustrated in Figure 2.
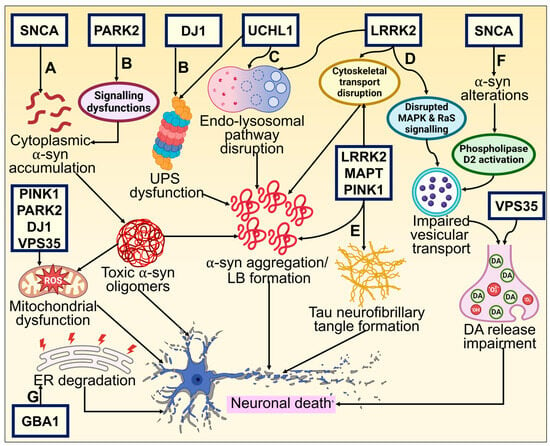
Figure 2. Genetic basis of PD and its underlying molecular pathways toward neurodegeneration. (A) The neuronal defense against α-syn aggregating could consequently be compromised by mutations altering these proteins. The cytoplasmic concentration of the α-syn monomer is increased by missense mutations and chromosomal multiplications of SNCA, promoting oligomerization of α-syn that is cytotoxic. This results in neuronal membrane damage and mitochondrial dysfunction [27,94]. (B) DJ-1 and Parkin (encoded by PARK2) interact and participate in regular UPS operations [95]. Mutations affecting these proteins may reduce the neuron’s ability to respond to α-synuclein aggregation. α-synuclein aggregates that build up inside neurites and axons in late-onset PD when there is residual UPS function eventually become trapped inside a central Lewy body in surviving neurons. DJ1 also possesses antioxidative characteristics, which may offer an additional connection to α-synuclein fibrillization and impaired function [96]. (C) UCHL1 preserves a pool of monoubiquitin for E3 ligase and UPS function while inhibiting the degradation of free ubiquitin in the endosomal–lysosomal pathway [97,98]. UPS functioning and protein buildup clearance need ATP generation by mitochondria. Loss of PINK1, DJ-1, and Parkin activities significantly impairs normal mitochondrial activity, which leads to early-onset parkinsonism [99]. (D, E) Tau (encoded by microtubule-associated protein tau—MAPT) typically maintains the microtubule network in equilibrium, facilitating intracellular signaling and neuronal trafficking. Abnormal phosphorylation impairs its functionality and causes neurofibrillary tangles to develop [100,101]. Phosphorylation, intracellular signaling, and cellular trafficking all seem to be interconnected events requiring LRRK2 [95]. (F) Mutation-induced altered α-syn activity causes reduced vesicular binding, which negates the inhibition of phospholipase D2, an enzyme implicated in vesicle trafficking and lipid-mediated signaling cascades [102]. Further, restricted release of neurotransmitters and their buildup in the cytosol may produce reactive oxygen species, which in turn causes neuronal death. (G) GBA1 mutation leads to ER stress activation and degradation, which causes DAergic neuronal death [103].
2. α-Synuclein
Gene multiplication and point mutation in the SNCA gene results in the autosomal dominant PD [104]. However, sporadic PD is believed to occur due to polymorphisms in the SNCA gene locus [105]. The SNCA/PARK1 gene encoding α-syn (A53T) was first recognized to cause a familial type of PD because of a missense mutation [106]. Various missense point mutations, namely, A53E, H50Q, A30P, G51D, and E46K, were also discovered shortly after that in the N-terminal region of α-syn. PD linked with SNCA frequently develops early and progresses quickly [107,108].
α-syn is a 140-aa protein that has three different domains: a strongly negatively charged C-terminal site, a hydrophobic non-amyloid domain that is capable of adopting a β-sheet conformity [109], and an N-terminus that takes on an α-helical secondary configuration over membrane binding. The protein structure and the positive and negative charges of α-syn are directly associated with pathological modifications of α-syn. The primary area for phosphorylation alterations is the negatively charged carboxyl terminus. By virtue of its hydrophobicity, the core hydrophobic region can easily form a β-pleated sheet. The positively charged amino terminus is vulnerable to acetylation and ubiquitination changes [110,111]. α-syn structural domains are depicted in Figure 3.
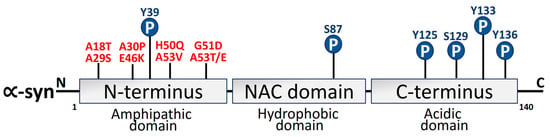
Figure 3. Schematic depiction of α-syn structure domains. Three different domains can be distinguished from the 140-amino-acid α-syn protein. The N-terminus amphipathic domain is composed of KTKEGV repeats that contain the amino acid residues affected by the primary α-syn gene mutations (A30P, E46K, H50Q, G51D, A53T, and A53E) in PD [112,113]. Mutations linked to Parkinson’s disease (red) and phosphorylation sites (blue) have also been depicted.
The CNS possesses high levels of α-syn expression, which is restricted to the area around synaptic vesicles that might be involved in synaptic transmission [114]. A study on the rat model of PD has shown that the α-syn variants that form oligomers tend to be more cytotoxic than those that form fibrils, resulting in an increasingly serious degeneration of DAergic neurons in the SNPC [115]. LB mainly consists of α-syn, which is phosphorylated at Ser129 of α-syn [116,117]. Amid pathological circumstances, α-syn may adopt a β-pleated secondary framework as the building block for LB and LN [118]. α-syn monomers assemble to generate protofibrils that resemble strings and may expand into bigger fibrils, further worsening the progression of PD pathogenesis [119]. According to certain research findings, oligomeric α-syn species that are toxic to neurons are compartmentalized by LB, suggesting that the aggregates of the protein may not pertain to neuronal toxicity [120,121]. The unfolded protein response (UPR) is provoked by SNCA gene triplication in a PD patient [122]. Matsui et al. [123] reported that T64 phosphorylation alters the characteristics of α-syn and promotes the generation of distinctive oligomers in the human PD brain. Such phosphomimetic mutation leads to lysosomal disorder, mitochondrial failure, neurodegeneration, and apoptosis, suggesting the pathogenic potential of α- syn phosphorylation at T64 in PD.
Protein post-translational modifications (PTMs) control protein activity and proteome modifications [124,125]. By changing the α-syn configuration, aggregating kinetics, subcellular localization, fibril ultrastructure, and molecular interactions, these PTMs considerably impact the emergence and dissemination of disease [126]. The combinational implications of PTMs, alongside related non-covalent cofactors on protein functioning, fibril organization, and pathological features, demand additional research, considering α-syn is susceptible to multiple alterations in both PD and non-PD individuals [127]. α-syn binds to the orexin 1 receptor (OX1R), which facilitates the post-translational protein degradation of OX1R through lysosomal and proteasomal pathways [128]. Extracellular signal-regulated kinase and protein kinase B signaling pathways are further downregulated, resulting in orexin neuron destruction that induces sleep behavior disorder, a potential early sign of PD [128]. The spectrin–ankyrin complex, crucial for the precise positioning and functionality of integral membrane proteins like Na1/K1 ATPase, is altered by the binding of α-syn to β-spectrin. Thus, neuronal dysfunction and mortality are caused by a higher level of α-syn in PD and associated α-synucleinopathies [129].
Genome-wide association studies revealed a link between PD and genetic variation in the gene for the tau protein, which is interlinked to AD [130] and regulates cytoskeletal integrity [131]. Interaction between α-syn and tau allows them to mutually promote each other’s aggregation [132]. In an α-syn overexpression model, hyperphosphorylated tau has also been identified [133]. In Drosophila, coexpression of α-syn increased the death of dopamine (DA) neurons induced by tau [134]. α-syn also seems to be involved in mitochondrial malfunction [135]. The chemical reaction of oligomeric α-syn with the membranes of mitochondria may result in their fragmentation and Dynamin-like protein 1 (DLP1)-independent mitochondrial fission [136]. Additionally, α-syn inhibits the functioning of mitochondrial complex I [137].
α-syn mutations potentially harm DAergic neurons, since they change several intracellular signal pathways. The mutations in α-syn A53T have the capacity to block autophagy in transgenic mice’s brains at an early stage and cause synucleinopathy [138]. Furthermore, in mouse models of PD, A53T causes apoptotic pathways in adrenal phaeochromocytoma (PC12) cells that are driven by endoplasmic reticulum (ER) stress and mitochondrial malfunction [139]. α-syn mutant overexpression in DA cells like PC12 and SH-SY5Y severely impairs proteasomal protein cleavage [140]. According to Matsumoto et al.’s [141] studies on CD-1 mice, erythrocyte-acquired extracellular vesicles with α-syn can pass the blood–brain barrier (BBB); this could be an entirely novel mechanism for the brain and peripheral nervous system to communicate during the onset and progression of PD. DAergic neurons die as a result of α-syn selective binding to tropomyosin receptor kinase B (TrkB) and the inhibition of the TrkB signaling pathway in the mouse model studies of PD [142,143].
Moreover, in mouse models of PD, A30P mutation could accelerate the degeneration of DAergic neurons by triggering microglia and increasing the phagocytic oxidase and macrophage-1 expression [139]. Decreasing the expression of α-syn may offer a viable treatment strategy, considering that both mRNA and protein levels increase twofold in mice models of PD [144,145]. Zharikov et al.’s [146] investigation of the rat model of PD stated that disruption of α-syn employing shRNA reduces the progression of motor impairments as well as DAergic neuron degradation. Further, the preclinical studies need to be validated by clinical investigations.
3. LRRK2
A total of 1–5% of sporadic PD and 5–13% of familial PD is associated with mutations in LRRK2 [147,148]. The LRRK2/PARK8 gene mutation, which results in autosomal dominant PD, carries the highest risk of familial PD [17,149]. I2020T, R1441G, G2385R, R1441C, R1628P, R1441H, G2019S, and Y1699C are seven of the documented missense LRRK2 variants that have been confirmed to be pathogenic. These mutations are found in various functional domains of LRRK2 [150,151]. It is fascinating to note that variations in LRRK2 seem to be population-specific [152,153,154,155,156,157,158,159,160]. LRRK2 is large in size with 2527 amino acids and is made up of multiple functional domains [161]. LRRK2 has two enzyme-like functions as its catalytic center, which comprises the Ras of complex (ROC) and the C-terminal of the ROC bidomain with the kinase region [162,163]. The structural domain representation of LRRK2 is given in Figure 4. LRRK2 is abundantly expressed in organs like the kidney, lungs, heart, and brain [164]. Additionally, it has been observed that LRRK2 can be identified in monocytes, lymphocytes, blood, cerebrospinal fluid (CSF), and urine [165]. Lipid dynamics are essential for vesicle trafficking, lipid metabolism, and lipid storage, all of which depend on LRRK2 substrates from the Ras-associated binding (Rab) GTPase family. Furthermore, LRRK2 is also linked to the phosphorylation and activity of enzymes that catabolize lysosomal lipids and the plasma membrane [166]. Elderly LRRK2 G2019S mutant carriers have substantially higher rates of PD morbidity [167].
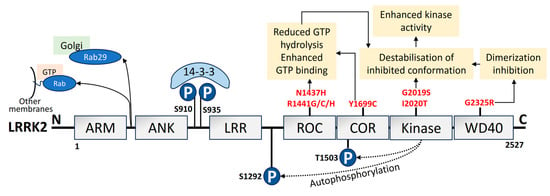
Figure 4. Domain organization, upstream regulation, and PD-linked pathogenic mutations of LRRK2. LRRK2 is made of 2527 amino acids. It consists of 7 domains: ARM, armadillo; ANK, ankyrin; LRR, Leucine-rich repeat; ROC, Ras of complex; COR, C-terminal of ROC; kinase; and WD40 domain [168]. The phosphorylation sites in the N-terminus are pSer910 and pSer935 (blue), which mediate 14-3-3 binding to LRRK2. The autophosphorylation sites are pSer1292 and pThr1503 (blue). PD mutations (red) lead to the pathological mechanism that increases kinase activity. Upstream regulation by LRRK2, such as the pathogenic VPS35 mutation, potential LRRK2 recruitment by unidentified Rabs to other organelle membranes or vesicles, and Rab29 recruitment to the Golgi has also been depicted [169].
In DAergic neurons and cultures of primary neuronal cells of a PD brain, LRRK2 G2019S mutation increases α-syn mobility and boosts α-syn accumulation [170]. The mutation has also been found to contribute to tau protein neural pathology in LRRK2-linked PD by promoting tau transmission in neuronal cells in mice [171]. LRRK2 interferes with DA signaling in addition to limiting neuronal survival. Through phosphorylation of apoptotic signal-regulating kinase 1 at the Thr832 site and boosting the kinase potential, LRRK2 performs significant roles in neuronal death [172]. LRRK2 has a variety of functions in the secretory pathway and might assist in DA signaling in a mice model of PD [173]. LRRK2 may cause neurons to die by suppressing myocyte-specific promoter factor 2D action, which is necessary for neural cell survival [174]. Dopamine receptor D1 uptake is compromised by the LRRK2 G2019S mutation, which alters signal transduction [175]. Additionally, the G2019S mutation increases kinase activity, impairing the synaptic vesicle transportation in ventral midbrain DAergic neurons [176]. A close correlation exists between mitochondrial dysfunction and the LRRK2 G2019S mutation. According to Howlett et al. [177], this mutation causes mitochondrial DNA (mt-DNA) destruction, which is reversible through a pharmacological decrease in the activity of LRRK2 kinase. Triggering mitochondrial DLP1 levels and neural toxicity, mitochondrial disintegration and dysfunction are caused by LRRK2 G2019S’s ability to bind to and strengthen its interaction with mitochondrial DLP1 [178,179].
4. PINK1
A 581-aa serine/threonine kinase is encoded by PINK1 [180]. The regions at the N-terminal oversee PINK1 processing and transport to mitochondria. The kinase domain consists of two lobes, N and C. A significant number of PD-linked mutations and well-characterized phosphorylation sites are in this PINK1 domain. PINK1 structural organization is depicted in Figure 5. The autosomal recessive PARK6 form of PD is brought on by PINK1 deficit [181]. Inner mitochondrial membrane-bound proteases cleave PINK1 at A103 and F104 to a 52-kD split, which escapes into the cytoplasm and is deteriorated by ubiquitination [182]. As a result, PINK1 basal levels are not detectable. Transport via the outer membrane of mitochondria is hampered by mitochondrial stressors like depolarization of the membrane, electron transport chain (ETC) unit malfunction, and mutagenesis stress, which stops proteolysis. This causes PINK1 buildup on the outer membrane of mitochondria, which triggers dimerization and activates the kinase domain [183]. As a result, PINK1 serves as a sensor for mitochondrial damage by turning on pathways for mitochondrial quality monitoring. Disruption of PINK1 and Parkin also causes an imbalance in the activity of the inositol 1,4,5-triphosphate receptor, which sharply increases calcium release from the ER [184].
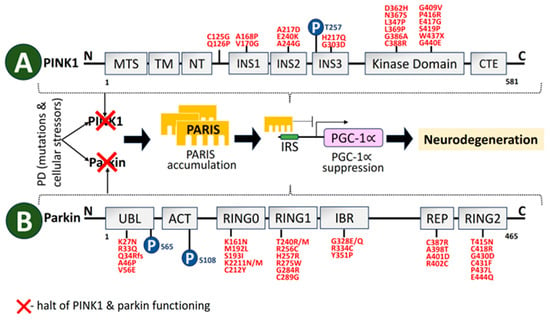
Figure 5. Schematic representation of structural domains of the mitochondrial-associated kinase PINK1, and the RBR-E3 Ubiquitin Ligase Parkin. (A) PINK1 is divided into distinct sections. Individual domains are depicted and labeled as follows: MTS, mitochondrial targeting sequence; TM, transmembrane domain; NT, N-terminal, regulatory domain; INS, insertion; CTE, C-terminal extension [185]. Depending on the residues and protein areas affected, PINK1-PD mutations (red) can be classified as having an impact on substrate binding, kinase activity, or PINK1 structure. (B) Parkin is comprised of 465 amino acids. Individual domains are depicted and labeled as follows: UBL, ubiquitin-like domain; ACT, activating element; RING, really interesting new gene domain; IBR, in-between-RING domain; REP, repressor element [186]. Mutations in PINK1 and Parkin cause PARIS accumulation that leads to neurodegeneration. Mutations linked to parkinson’s disease (red) and phosphorylation sites (blue) in PINK1 and parkin have also been depicted.
By phosphorylating LETM1 at Thr192 to promote Ca2+ release while facilitating its transportation, PINK1 depletion is connected to mitochondrial failure and mitochondrial Ca2+ dysregulation [187]. According to Martinez et al. [188], the deregulation of the misfolded protein response of mitochondria relying on the PINK1 homolog causes non-apoptotic degeneration of DAergic neurons. A dominant-negative pathway may raise the risk of PD in heterozygous PINK1 G411S mutation carriers, as this mutation drastically reduces PINK1 kinase function [189]. In PD mice, PINK1 mutations promote the buildup of defective mitochondria with aging, stimulating the misfolded protein response of mitochondria and prolonging life [190]. The PINK1 mutations I368N and Q456X decrease either protein stability, levels, or kinase activity, raising the likelihood of PD [191]. Parkin is less likely to be recruited to depolarized mitochondria by other mutations, such as G309D, A168P, L347P, and H271Q [192].
5. Parkin
Originally thought to function as ubiquitin E3 ligase that could be stimulated by autophosphorylated PINK1 [193], Parkin is a protein that is encoded by PARK2 and is necessary for the degradation of target molecules through the ubiquitin–proteasome system [194]. The structural domains of Parkin are shown in Figure 5. A total of 10–25% of early-onset PD occurs due to Parkin gene mutations [195]. In order to facilitate the mitophagy destruction of defective mitochondria, PINK1 aggregates on the malfunctioned mitochondrial membrane stimulate Parkin E3 ubiquitin ligase operation and recruit cytoplasmic Parkin molecules to the dysfunctional mitochondria [196]. By encouraging Parkin recruitment to mitochondria, reactive oxygen species (ROS) also trigger PINK1/Parkin pathway-controlled mitophagy [197]. A defect in Parkin recruitment for depolarizing mitochondria results from mutations such as C441R, R42P, C289G, R46P, C253Y, C212Y, and K211N, which substantially restrict mitophagy [198]. Mitophagy and mitochondrial biogenesis are coordinated by the Parkin interacting substrate (PARIS) axis. A network of sound mitochondria is maintained by basal state cellular homeostasis. A transcriptional program involved in mitochondrial biogenesis is connected to mitophagy. Parkin and PARIS are involved in one pathway in this intricately regulated process. The steady-state levels of PARIS are regulated by Parkin-mediated ubiquitination, which is aided by PINK1 as a priming kinase. Parkin expression and activity are increased by mitophagy triggers, which causes PARIS to be broken down by proteases. Reduced levels of PARIS alleviate Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) transcriptional inhibition, hence facilitating mitochondrial biogenesis [199].
6. DJ-1
Early onset of recessive PD has been linked to mutations in DJ-1, which is encoded by the PARK7 gene [200]. A PD-linked gene retained in both prokaryotes and eukaryotes, DJ-1 (PARK7), is an evolutionary ancient gene [96,99,201]. The functional domains of DJ-1 are shown in Figure 6. The function-related mutational loss that affects DJ-1 protein integrity and homo-dimerization causes PARK7 PD, which is inheritable in an autosomal recessive manner [202]. Several DJ-1 mutant variants have been linked to PD, including L166P, M26I, L10P, and P158. It has been reported that a DJ-1-associated PD brain has neuropathological issues as per the genetic data, indicating a distinctive mutation of L172Q in the PARK7 gene [203]. In cells overexpressing PINK1, DJ-1 might interact with and stabilize PINK1 [204]. DJ-1 also engages with α-syn, which modifies its accumulation by establishing a weak hydrophobic interaction [205] and reversing α-syn-mediated toxicity to cells [206]. Along with PARP1, DJ-1 preserves genomic stability, and disruption of this connection might have an influence on DNA damage accumulation, impaired DNA repair, and, ultimately, neurodegeneration. Therefore, faulty DNA repair is associated with the PD pathophysiology brought on by DJ-1 mutations [207].
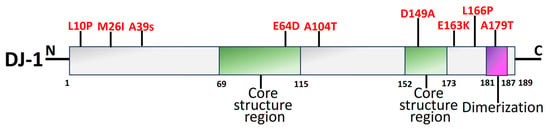
Figure 6. An illustration of functional domains of the DJ-1 protein with pathogenic mutations (red). The two core structural areas (green) and the dimerization region (purple) make up the DJ-1 protein. DJ-1 is a single-domain protein with 189 amino acids [208].
The antioxidant capabilities, antiapoptotic implications, and impact on mitochondrial respiration, shape, shifts, and biosynthesis are a brief overview of the neuroprotective actions exerted by DJ-1 [209]. DJ-1 mutations principally affect a protein that facilitates intracellular oxidation–reduction [210]. Due to its elimination of neurological protective effects against H2O2 and elevation of thioredoxin-1 by suppressing the nuclear factor erythroid 2-related factor 2 signal pathway, mutant DJ-1 (L166P and M26I) raises the vulnerability of SH-SY5Y cells to oxidative stress [211]. Additionally, the D149A mutation ends this shielding [212]. L172Q, L10P, and P158∆ are the three mutations that contribute to a decrease in the stabilization of proteins [213]. In vivo experiments have demonstrated that recombinant DJ-1 can also stop the DA degeneration caused by 6-hydroxydopamine (6-OHDA) or α-syn [214].
7. Vacuolar Protein Sorting 35 (VPS35)
The endolysosomal network is made up of many tubulovesicular organelles that are important for protein manufacturing, nutrition intake, cellular trafficking, and apoptosis [215,216]. The heterotrimeric complex, comprising VPS26, VPS29, and VPS35, is a crucial component of the endolysosomal system’s sorting process [217,218,219]. VPS35 structural domains are represented in Figure 7. The mechanism of transmembrane protein shifting between Golgi and endosomes is regulated by VPS35 [220,221]. The VPS35 gene was originally linked to late-onset PD in an Austrian family. It contributes 1% to familial PD. The VPS35 D620N mutation is harmful in PD patients from American, European, and Asian families [222]. In PD-afflicted fibroblasts, the VPS35 D620N mutation decreases enzyme functionality in complex I and II, resulting in mitochondrial failure by reusing DLP1 complexes [136,223]. Finally, DAergic neuron loss occurs as a result of mitochondrial malfunction brought on by VPS35 deficiency [224]. PD-related VPS35 mutation, R524W, hinders retromer endosomal interaction and causes α-syn to aggregate [225]. In a Drosophila model, the VPS35 P316S mutation also caused some PD symptoms, such as decreased climbing power and a reduction in Daergic neurons, which made the fly particularly susceptible to the drug rotenone [226].
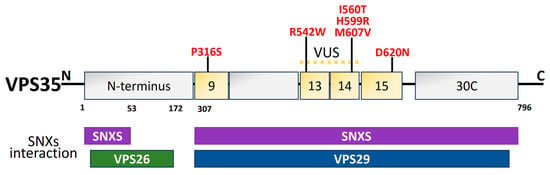
Figure 7. Schematic illustration of VPS35 structural domains and interactions along with the PD mutations (red) [208]. The dimer of sinexin (SNXs) and VPS26, VPS29, and VPS35 combine to form the retromer cargo recognition complex. For the interaction with VPS26 and VPS29, the amino acid residues 1–172 in the N-terminal region and 307–796 in the C-terminal region are significant. The N- and C-terminal regions of the amino acid residues are those that interact with the SNXs. Thirteen of the thirty-four helices projected for a structural level VPS35, a right-handed α-helix solenoid, are predicted to be in the C-terminal. In PD, several missense mutations have been discovered. The location of the VPS35 variation of unknown significance (VUS) is between exons 9 and 14. Exon 15 is the site of the pathogenic mutation (D620N).
8. Glucocerebrosidase 1 (GBA1)
The 497-aa protein β-glucocerebrosidase (GCase1) is encoded by GBA1, which is located on the lysosomal membrane. GBA1 structural domain organization is illustrated in Figure 8. The most prevalent gene-associated PD risk factor is the catabolism of the glycolipid glucocerebroside into glucose and ceramide in the lysosome [227,228], which causes Gaucher disease. In 7–12% of patients, heterozygous GBA1 mutations have been identified as the most prevalent genetic risk factors for PD [229]. As patients with PD or GBA1 mutations may display extensive LB or LN, GCase1 malfunction and α-syn pathogenesis appear to be related inextricably [121]. In addition, GCase1 enzyme activity is decreased in GBA1-PD and sporadic PD cases [230,231] and has an inverse relationship with the degree of α-syn pathology [232].
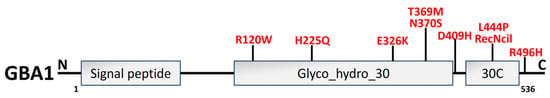
Figure 8. An illustration of GBA1 structural organization along with PD mutations (red). GBA1 protein is composed of 497 amino acids, which have three primary domains: 39-residue signal peptide, the conserved catalytic domain Glyco_hydro_30 (329 amino acids), and Glyco_hydro_30C domain (30C; 62 amino acids) [233].
In the brain and CSF of PD patients, an enormous decline in GCase 1 functioning and protein concentrations has also been identified [234,235]. Employing a small-molecule modulator to activate GCase 1 reinstated lysosome functionality and eliminated the buildup of pathogenic α-syn in PD individuals, suggesting the plausible role of GCase 1 in the onset of idiopathic PD [236]. According to estimates, L444P and N370S are the two most prevalent GBA1 mutations, accounting for around 10% to 25% of PD instances [237,238]. The instability of α-syn tetramers is reversed, and human DAergic neurons are protected against α-syn prefabricated fibril-initiated toxicity by inhibiting glycosphingolipids accumulation and overly expressed GBA1 to increase GCase 1 function [239]. The mutation of N370S caused GCase 1 to be retained in ER, stopping its flow to the lysosome, which activated the UPR and caused the Golgi apparatus to fragment by ER stress activation [240,241]. In addition, heterozygous L444P GBA1-mutated murine neurons showed dropped α-syn tetramers along with associated multimers [239], and L444P GBA1 mutation provoked α-syn-mediated DAergic neuronal loss in the SNPC of mice models of PD [242].
This entry is adapted from the peer-reviewed paper 10.3390/biom14010073
This entry is offline, you can click here to edit this entry!