Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder impairing cognition and memory in the elderly. This disorder has a complex etiology, including senile plaque and neurofibrillary tangle formation, neuroinflammation, oxidative stress, and damaged neuroplasticity. The treatment options are limited, so alternative treatments such as herbal medicine could suppress symptoms while slowing cognitive decline.
- Alzheimer’s disease (AD)
- kaempferol
- quercetin
- flavonoids
- traditional Chinese medicine
- dementia
1. Introduction
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder characterized by cognitive decline and memory impairment. AD could affect 152 million individuals by 2050 [1]. The progression of AD is influenced by multiple factors, including the accumulation of beta-amyloid plaques (Aβ) and the formation of neurofibrillary tangles (NFTs). The aggregation of Aβ plaques exacerbates the disease by impairing neuronal function and triggering neuroinflammation [2,3,4,5]. Oxidative stress and the presence of neurofibrillary tangles (NFTs) also contribute to the aggregation of Aβ into senile plaques [6,7,8,9,10,11,12,13,14]. NFTs consist of hyperphosphorylated tau proteins that disrupt neuronal transport systems [15,16,17,18,19,20,21]. Neuroinflammation, in turn, exacerbates damage to neuronal integrity [22]. Symptoms of AD include memory loss, impaired learning, emotional changes, cognitive and speech deficits, shortened attention span, and impaired management of daily tasks [23,24,25].
Currently, the treatments available for AD are expensive and have minimal efficacy. Acetylcholinesterase inhibitors (AChEIs), including donepezil, and N-methyl-D-aspartate (NMDA) receptor antagonists, including memantine, are commonly prescribed for AD [26]. AChEIs inhibit the enzymatic degradation of ACh by inhibiting cholinesterase activity [27], while NMDA receptor antagonists limit calcium influx to prevent glutamate-induced cytotoxic cell death [28]. However, these drugs simply suppress symptoms and fail to halt disease progression [26], and only half of the population positively responds to these current treatments [29,30]. Herbal medicine boasts a well-documented history of safe and effective incorporation into traditional Asian diets [31,32]. Preclinical studies have demonstrated that these herbs can enhance cognitive and memory functions [33,34]. These herbs serve as dependable sources of phytochemicals, such as kaempferol and quercetin, that have limited side effects and could combat Alzheimer’s disease [35,36,37]. Specifically, these flavonoids have anti-inflammatory, neuroprotective, and anti-degenerative effects [33,38,39,40,41,42,43,44,45,46].
2. Hallmarks of Alzheimer’s Disease
Several features of AD, including Aβ plaque accumulation [47,48], tau hyperphosphorylation and neuroinflammation [49], and oxidative stress [50,51,52,53], have been identified as targets for drug development. Moreover, these deficits have been observed in studies with human patients [6,54,55,56,57,58,59,60,61,62,63]. This section will briefly explore the pathophysiology of AD, with a focus on the proposed molecular origins and outcomes of their aberrant activities.
While the origins are still debated, the literature greatly supports the roles of oxidative stress and neuroinflammation as critical drivers of neurodegeneration. Antioxidant deficits facilitate ROS production, driving oxidative stress via lipid peroxidation [57]. Consequently, mitochondrial energy production is impaired and pro-apoptotic signaling follows [57]. Glutamate-induced excitotoxicity could also facilitate oxidative stress [64,65,66,67]. Disrupted ROS clearance establishes the neuroinflammatory microglial and astrocytic hyperactivity [38,68,69,70,71,72] and favors neuronal signaling pathways that impair Aβ clearance [48,73,74]. Finally, proper mitochondrial function is required for Aβ clearance and can, in turn, maintain appropriate tau activity states [75].
Although normal Aβ levels can maintain regular neuronal function [76], failed Aβ clearance from the brain can expedite neurodegeneration by facilitating plaque accumulation and impairing neuronal communication [22,47,48,75,77,78,79]. Moreover, Aβ accumulation further promotes oxidative stress [80,81,82,83]. As Aβ plaques accumulate in the brain due to impaired clearance [84], overzealous astrocytic and microglial responses compound the neuroinflammatory environment by releasing pro-inflammatory factors, promoting neuronal apoptosis [6,49,85,86,87,88,89]. These findings were supported in postmortem tissue [6,54,55,56]. Finally, Aβ signaling significantly impairs LTP [90], facilitating neurodegeneration via low synaptic activity.
AD is one of the most common tauopathies [91]. Aβ plaque accumulation drives tau hyperphosphorylation [47,58,92,93,94,95,96,97,98,99,100], possibly by excess GSK-3β signaling [101]. Likewise, tau hyperphosphorylation also compounds Aβ toxicity [102,103], which has been supported by PET imaging in humans with memory impairment and cognitive decline [60]. These studies demonstrate that Aβ toxicity is necessary for tau hyperphosphorylation [59,60]. Specifically, accumulating Aβ binds to NMDAR, generating excess calcium levels to activate calpain-mediated microtubule-associated protein cleavage [65,104,105]. These events impair mitochondrial function, invoking pro-apoptotic signaling [65,106]. Tau hyperphosphorylation dismantles axonal microtubules to degenerate the axon [15,107,108,109,110], impairing synaptic plasticity [102,103,111,112]. Hyperphosphorylated tau spreads throughout the hippocampus in AD models [113], and uptake may be mediated by clathrin-induced endocytosis [114]. Risk factors such as sleep apnea may potentiate the spread of tau in this manner [115]. Ultimately, these events result in neuronal death and compromise neuroplasticity, thereby driving neuroinflammation and impairing cognitive function.
3. Anti-AD Mechanisms of Quercetin and Kaempferol
Given the limited therapeutics available to AD patients, it is essential to explore alternative treatments, such as plant-derived phytochemicals. Flavonoids, including kaempferol and quercetin, belong to the class of polyphenols commonly found in various herbs. Notably, kaempferol and quercetin possess lipophilic properties [50], which facilitate their easy entry into cells. These phytochemicals are abundant, with an average daily consumption of approximately 23 mg of flavonoids in a typical diet [116,117]. Kaempferol and quercetin produce several beneficial properties, including anti-inflammatory, antioxidant, anti-Aβ, anti-tau, and pro-neuroplastic effects [37,38,39,57,74,118,119,120,121,122,123,124,125,126,127,128]. Moreover, they have demonstrated cognitive and memory-enhancing effects in animal studies [37].
3.1. Quercetin
Quercetin, the most prevalent flavonoid, is found in several traditional medicinal herbs and is commonly found in fruits and vegetables, including berries, onions, and leeks [118,129,130,131,132,133,134,135,136,137,138,139]. Quercetin intake constitutes approximately 60–75% of total flavonols [140,141], and 25 mg of quercetin is found in the average diet [38]. Quercetin is commonly investigated for its potential anti-neurodegenerative efficacy and is considered safe [51,142]. Quercetin is a 15-carbon flavonoid with two benzene rings connected via a 3-carbon shape (Figure 1) [38,130,143].
Quercetin produces anti-inflammatory effects via multiple signaling pathways, including Nrf2, paraoxonase-2 (PON2), JNK, PKC, and NF-kB [51,118,128,144,145,146,147]. Quercetin dose-dependently protected HT22 hippocampal neurons from glutamate-induced apoptosis by limiting ROS production, impairing the calpain-mediated cleavage of cytoskeletal proteins, and preserving mitochondrial membrane potential [65]. Quercetin also inhibits NO release by inhibiting iNOS activity [33,38,148], which could reduce excess glutamate signaling and minimize the risk of glutamate-induced cytotoxicity in hippocampal neurons in a similar fashion to kaempferol and its derivatives [149]. Moreover, quercetin inhibits COX-2 and TLR4 activity to reduce inflammatory responses [6,39,148]. Interestingly, quercetin may have epigenetic mechanisms by inhibiting lysine acetyltransferase (KAT) activity [150,151] and increasing lysine deacetylase (KDAC) activity [152], suggesting that the flavonoid can bidirectionally regulate autophagy [153], neuroinflammation, and apoptosis [154]. Quercetin also inhibits acetylcholinesterase (AChE) [155], which can enhance alertness and cognitive function in AD patients.
The anti-Aβ effects of quercetin are well studied in AD and related models and have yielded promising therapeutic properties. The hydrophobic groups of quercetin can inhibit the formation of Aβ fibrils [120,121,122,123,156]. Chronic quercetin treatment also slowed Aβ aggregation by potentiating AMPK signaling and inhibiting mitochondrial ROS production, leading to improved memory and object recognition in APPswe/PS1dE9 [80,157]. Quercetin treatment also inhibits the BACE1-mediated cleavage of APP into Aβ by inhibiting NF-kB [74]. Consequently, mitochondrial membrane permeability is restored, and cellular survival is favored over oxidative stress [158]. This anti-neurodegenerative effect could be due to the free radical-quenching structure of the catechol group, reducing neuroinflammation, lipid peroxidation, mitochondrial stress, and DNA damage [38,51]. Elevated SOD, GPx, and Na+-K+ ATPase activity could also be due to quercetin’s anti-Aβ effects [44,78].
In many studies, quercetin and its derivatives reduced tau hyperphosphorylation [23,58,132,159]. In rodent HT22 hippocampal neurons, chronic quercetin treatment inhibited tau phosphorylation at four sites by reducing p-Cdk5 levels, limiting calpain activity, and dramatically reducing Ca2+ influx [58]. In 3xTgAD mice, chronic quercetin inhibited Aβ pathology, reduced NFT levels, and prevented astrocytic and microglial hyperactivity in the amygdala and hippocampus [132,160], showing that the anti-Aβ and anti-tau mechanisms of quercetin depend on its anti-inflammatory effects. Consequently, these mice demonstrated improved learning and memory and decreased anxiety [132], while combined exercise and quercetin treatment robustly improved spatial memory in AD rodents [161]. Studies also found that quercetin enhanced cell viability and morphology by reducing MDA and ROS levels and increasing antioxidant SOD and GSH activity [159,162], limiting NF-κB signaling, restoring mitochondrial membrane potential to baseline, inhibiting tau hyperphosphorylation, and regulating Akt/PI3K/GSK-3β signaling pathway [159,163]. Taken together, these data show that quercetin has a multimodal mechanism of action in treating AD. Of note, the anti-tau and consequent pro-neuroplastic effect of quercetin is further explored in Section 5, but the primary anti-inflammatory, anti-Aβ, ant-tau, and pro-neuroplastic effects of this flavonoid are all dependent on each other.
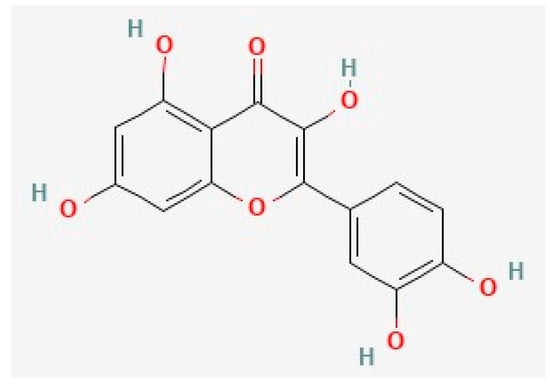
Figure 1. The chemical structure of quercetin, deduced from PubChem [164].
3.2. Kaempferol
Kaempferol is a common 15-carbon polyphenol (Figure 2) that shares significant structural similarity with quercetin. It is one of the most common flavonoids and is found in a variety of common foods, including fruits and vegetables [129,130,165,166,167,168,169,170]. Multiple preclinical and clinical studies have supported the anti-AD activity of kaempferol [57,149,171,172,173,174]. Kaempferol has pro-neuroplastic, anti-Aβ, anti-tau, anti-inflammatory, and antioxidant properties [29,44,57,171,175,176,177,178,179]. Notably, kaempferol also inhibits AChE like quercetin [180], but this mechanism is beyond the scope of this review.
Like quercetin, kaempferol and its metabolites reduce inflammation and have potent antioxidant properties [181,182,183,184]. Kaempferol also directly modulates neuroinflammation by impairing microglial TLR4 and NF-kB signaling and inhibiting the release of NO, iNOS, PGE2, IL-1β, TNF-α, and IFN-γ [167,185]. Kaempferol also reversed BBB damage [36,186,187]. Kaempferol can also modulate neuroinflammation by regulating epigenetic factors such as SIRT1, a subtype of KDAC [188,189,190]. Kaempferol also prevents cytotoxic damage to PC12 neurons by upregulating SIRT [191]. Other immune factors modulated by kaempferol include COX-2, lipoxygenases, prostacyclin, and leukotrienes [148,187,192,193,194]. Finally, kaempferol may reduce neuroinflammation via Nrf-2 signaling [185].
Like quercetin, kaempferol and its derivatives reverse Aβ-induced damage [29,120,122,124,125,149,195]. Kaempferol-3-O-rhamnoside (K-3-Rh), a kaempferol derivative, limited total Aβ burden and toxicity by disrupting β-sheet formation and impairing Aβ plaque formation in human SH-SY5Y cells [195,196]. However, kaempferol antagonized fibrilization with lower potency compared to quercetin and morin [120,122]. In rodent neuroblastoma cells, kaempferol 3-O-(6″-acetyl)-β-glucopyranoside (KAG) robustly inhibited Aβ-mediated cytotoxic cell death and ROS generation [149]. KAG reversed Aβ-mediated oxidative stress and increased cell survival by regulating caspase-3, Bax, and Bcl-2 signaling [44,64,149,197,198,199,200]. Kaempferol dose-dependently and sex-dependently limited Aβ-induced mitochondrial toxicity in neurons, improving rodent memory in the Y-maze test [57,134,201]. Of note, studies regarding kaempferol’s direct influence on tau are limited; thus, more research is necessary. However, due to its similar phenolic structure to quercetin [165,166], we hypothesize that kaempferol could also reduce tau hyperphosphorylation.
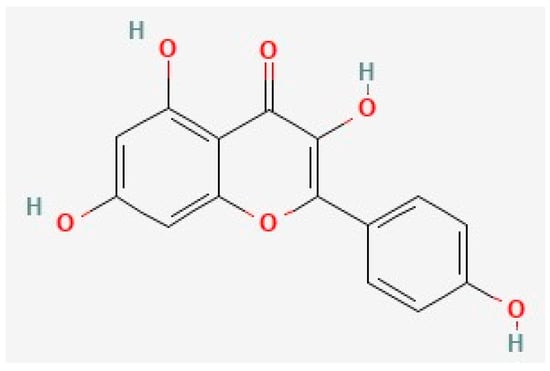
Figure 2. The chemical structure of kaempferol, deduced from PubChem [202].
4. Kaempferol, Quercetin, and Neuroplasticity
The aberrant brain changes described in Section 3 can impair memory and cognitive function by creating deficits in neuroplasticity. Thus, future AD treatments should also be designed to directly target signaling pathways that can counteract the etiologies of AD. Specifically, we identified the PI3K/AKT signaling pathway as a critical candidate to counteract neurodegeneration. Several studies have suggested that flavonoids can alleviate learning and memory deficits by targeting this signaling pathway [29,203,204,205]. However, other pathways, including the MAPK-ERK1/2 cascade [206], have also been proposed and outlined in a recent review [207].
4.1. Neuroplasticity Deficits in AD
An ideal AD treatment should enhance the expression of plasticity-related genes such as BDNF, a neurotrophic factor that regulates neuronal plasticity and survival [208,209,210,211,212,213,214]. BDNF signaling begins with its binding to the receptor, Trkβ, activating signaling via a variety of pathways like PI3K/AKT [211,215]. Then, AKT or protein kinase B (PKB) [216] can activate the CREB-mediated transcription of BDNF [217,218]. Since Trkβ receptors mediate the pro-neuroplastic effects of BDNF [219], AD drugs must produce a direct or indirect effect on the receptor.
BDNF deficits increase the risk of AD development [220], and BDNF dysfunction due to impaired PI3K and AKT signaling can expedite neurodegeneration [7,41,221,222,223]. The PI3K/AKT signaling pathway has multiple functions, including regulating synaptic plasticity, glucose processing, cell cycle progression, cell proliferation, survival, and apoptosis [167,175,224,225,226]. Moreover, this pathway may protect neurons from Aβ toxicity [224], oxidative stress [227], and neuroinflammation [217]. GSK-3β is downstream of PI3K/AKT, and Aβ can specifically lead to its hyperactivity [7]. However, BDNF and CREB are also vulnerable to Aβ signaling [228] as CREB is regulated by the PI3K/AKT/GSK-3β pathway [211,212,215,229,230,231].
Thus, the Aβ-mediated signaling cascade that degenerates the neuron is as follows (Figure 3A): Aβ binding to NMDAR inhibits PI3K/AKT signaling by activating GSK-3β-mediated tau hyperphosphorylation and CREB downregulation [93,97,210,211,223,229,230,232,233,234,235,236,237]. Consequently, the impaired CREB-mediated transcription of BDNF genes decreases plasticity and facilitates plaque accumulation, as demonstrated in postmortem tissues from humans and human neuronal cells [209,210,229,232]. The absence of protective BDNF and PI3K/AKT activity facilitates the caspase-mediated pro-apoptotic signaling cascade [6,224], degenerating the neuronal circuitry, while tau dissociation from microtubules breaks down the neuronal cytoskeleton [7,233,238,239,240,241,242].
4.2. Quercetin and Kaempferol Resolve AD-Related Plasticity Deficits
The multimodal mechanisms of kaempferol and quercetin collectively slow neurodegeneration by combating the impairments that are illustrated in Figure 3A. Specifically, the restoration of proper PI3K/AKT signaling will greatly improve synaptic plasticity deficits in AD [7]. While quercetin’s interaction with each component of this signaling pathway has already been documented [7], kaempferol’s mechanisms are still unclear. However, since kaempferol’s structure is similar to that of quercetin [165], we propose that kaempferol has a nearly identical mechanism with respect to the signaling pathway in this subsection. Finally, we will propose the potential outcomes of these molecular interactions.
Molecular docking studies suggested that quercetin can bind PI3K, AKT, and GSK3β [213,250,255,256,257,260,264,265]. Specifically, quercetin can bind to PI3K [256], consequently activating AKT signaling [265], or quercetin can directly bind to AKT [257]. In preclinical studies, quercetin reduced GSK-3β activity, which decreased tau hyperphosphorylation and reduced pro-apoptotic signaling [7,38,159]. Quercetin treatment in rodents also increased BDNF, Trkβ, PI3K, and AKT expression [243,266]. Consequently, quercetin enhanced neurite outgrowth in hippocampal neurons [36] and ameliorated the stress-induced downregulation of CREB and BDNF [40], suggesting that quercetin could potently replenish neuroplasticity in the AD brain. Moreover, quercetin inhibited Aβ by restoring Trkβ signaling and CREB-mediated BDNF transcription, increasing the viability of SH-SY5Y cells [252]. Finally, quercetin’s dual pro-neuroplastic and anti-inflammatory effects may also be related to the quercetin-mediated downregulation of BACE1 expression via the inhibition of NF-kB [253,254,264,267]. Taken together, these data suggest that quercetin antagonizes Aβ-induced GSK-3β signaling relative to tau by activating the PI3K/AKT pathway and directly inhibiting GSK-3β [7,225,241,255,256,260]. Consequently, proper BDNF levels can be restored to replenish neuronal plasticity in the AD brain. Similar chemicals, such as epigallocatechin-3-gallate (EGCG), attenuated tau hyperphosphorylation in a similar mechanism [23,268,269,270]. Thus, quercetin clearly has dual neuroprotective and pro-neuroplastic mechanisms in cells [33,65,252], and the clinical outcomes of quercetin’s pro-neuroplastic mechanisms were supported by its memory and cognition-boosting effects in rodent models of AD and Parkinson’s disease [23,38,44,271,272,273,274,275,276].
First, kaempferol improved hippocampal plasticity following traumatic brain injury in young rodents [277] and improved memory in rodents [29,57] and Drosophila [173]. Moreover, kaempferol dose-dependently maintained cell viability following Aβ treatment in multiple studies [29,149,195,248]. This could be due to kaempferol’s inhibition of BACE1-mediated Aβ synthesis [253,254] or the activation of the PI3K/AKT signaling pathway, enhancing CREB-mediated BDNF transcription [175,211,258]. Although one molecular docking study suggested that kaempferol may have minimal affinity for GSK-3β [250], kaempferol likely inhibits GSK-3β indirectly by first binding and activating PI3K [256] or AKT [175,185,257]. Via this mechanism, kaempferol prevents tau hyperphosphorylation, protecting neuronal morphology and function [47,278,279,280,281]. Then, AKT can activate CREB-mediated BDNF transcription [217]. Supporting this pro-neuroplastic mechanism, kaempferol and its metabolite, kaempferide, produced similar effects that resulted in Trkβ signaling [171,210] and enhanced BDNF expression in Aβ-treated mice [243]. Taken together, these data suggest that kaempferol enhances neuroplasticity to reverse Aβ damage by activating the PI3K/AKT cascade, which potentiates CREB-mediated BDNF transcription. However, kaempferol produces the opposite effect on this signaling pathway in microglial cells [167] and cancer cells [282]. Thus, kaempferol’s effects on the PI3K/AKT signaling cascade are dynamic and depend on cell lineage.
Despite the lack of literature demonstrating a direct modulation of tau by kaempferol, there is plenty of evidence to support the possibility that kaempferol inhibits tau hyperphosphorylation via the PI3K/AKT pathway and by antagonizing Aβ-mediated GSK-3β signaling [29,149,195].
These data suggest a clear anti-AD mechanism of quercetin and kaempferol, as outlined in Figure 3B. First, quercetin and kaempferol could enter the cell cytoplasm due to their lipophilic polyphenolic structure. Quercetin and kaempferol scavenge ROS and activate PI3K/AKT signaling to inhibit GSK-3β. Specifically, they can bind directly to PI3K or AKT to activate protective signaling, inhibiting GSK-3β and preventing tau hyperphosphorylation. This signaling cascade reduces the formation of NFTs in the AD brain. GSK-3β inhibition can also antagonize Aβ-NMDAR interactions. Thus, downstream pro-apoptotic signaling mediators are also inhibited by quercetin and kaempferol treatment. Due to reduced NFT and amyloid plaque formation, microglial hyperactivity decreases in the absence of the burden of clearance. Thus, progressive neuroinflammatory signaling is slowed, allowing surrounding neuronal synapses to survive. After chronic quercetin treatment, progressive elevations in BDNF release rebuild damaged synapses by favoring neurotrophic signaling over cytotoxic Aβ signaling, improving memory and cognition. Of note, molecular docking studies have not supported the possibility that kaempferol and quercetin can directly bind to tau protein, supporting their indirect inhibitory mechanism via GSK-3β inhibition. Taken together, kaempferol and quercetin share multiple mechanisms that slow AD progression by first limiting ROS activity, NFT aggregation, and Aβ-mediated toxic signaling, slowing neurodegeneration.
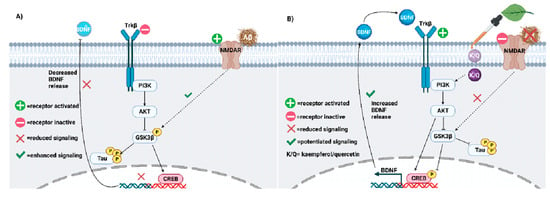
Figure 3. (A) Neuroplasticity deficits accelerate AD progression and must be treated. Impaired PI3K-AKT signaling facilitates GSK3β-mediated phosphorylation of tau. Aβ may potentiate tau hyperphosphorylation via GSK3β. (B) Kaempferol and quercetin (K/Q) invoke the PI3K/AKT pathway to antagonize Aβ and reduce tau hyperphosphorylation in neurons. As a result, neuroplasticity is increased in the AD brain [283].
5. Quercetin and Kaempferol in Common Herbs
Although data on the co-treatment of quercetin and kaempferol are still somewhat limited, the abundance of both compounds in several common herbs requires the investigation of the synergistic effects of both flavonoids, in addition to their interactions with other herbal phytochemicals. Flavonoid-rich herbs are commonly employed in traditional Chinese medicine (TCM), in which an emphasis is placed on the utility of natural treatments. Moreover, these herbs are generally safe for consumption [224]. Kaempferol is the second most common flavonoid in traditional medicinal herbs, following quercetin [225,284]. Other reviews have assessed the efficacy and safety of natural medicine in the treatment of neurodegenerative diseases [7,224], highlighting the potential medicinal properties of herbs in treating AD. Flavonoids are commonly found in herbs such as Schima wallichii Korth, Maesa membranacea, Ginkgo biloba, and many more [175,225,278]. These phytochemicals could work synergistically with each other and with other herbal components to invoke anti-AD effects. Thus, we explore common herbal sources of kaempferol and quercetin, describe the anti-AD mechanisms of herbs, and propose a design for a future AD treatment based on the current evidence of these effects.
Ginkgo biloba is a quercetin- and kaempferol-rich herb proposed to treat AD [285]. G. biloba improves memory and cognition by inhibiting ROS, facilitating hippocampal neuron proliferation, halting Aβ plaque accumulation, and reducing tau hyperphosphorylation [47,286,287,288]. Moreover, this effect is associated with reduced GSK-3β activity and the increased expression of PSD-95 and synapsin-1 [47]. As seen with kaempferol and quercetin alone, G. biloba potentiates PI3K/AKT relative to CREB signaling to promote neuroplasticity [287,289,290,291,292,293]. Hippophae rhamnoides extracts are also rich in quercetin and kaempferol, and they enhanced neuronal differentiation and neurite outgrowth via PI3K/AKT and ERK signaling [294,295]. However, clinical trials have revealed the inconsistent efficacy of G. biloba on cognition and other AD-related parameters [296]. Camellia sinensis is another kaempferol- and quercetin-rich herb commonly grown to produce black and green tea [297,298]. C. sinensis extracts improved spatial memory and reduced hippocampal Aβ fibrillization in AD rodents and had greater antioxidant effects compared to other herbs [298,299]. Kaempferol and its derivatives are found in the leaves of Maesa membranacea, Schima wallichii Korth, Carthamus tinctorius, Panax ginseng, and several other herbs [175,188,225,278]. S. wallichii was neuroprotective due to the promotion of hippocampal and cortical AKT signaling [175], and M. membranacea could protect H202-treated SH-SY5Y cells [225] and hippocampal tissue [300] via the same pathway due to their kaempferol abundance. C. tinctorus is rich in kaempferol, produces a similar effect, and invokes protective AMPK signaling [188]. Finally, recent studies also suggested that other herbs such as Morenga oleifera, Cuscuta chinensis, Allium cepa, Litchi chinensis, Prakia roxburghii, Radix astragali, Acoritatan Fagopyrum tataricum, Carthami flos, Punica granatum, and Cyperi rhizoma [251,257,264,301,302,303,304,305] may also be great sources of kaempferol and/or quercetin and produce anti-AD effects.
Polyherbal cocktails, such as Chaihu shugan san (CSS) and Huangqi Sijunzi (HQSJDZ), could treat AD and its risk factors. CSS is abundant in kaempferol and quercetin and contains herbs such as Glycyrrhiza uralensis, Cyperus rotundus, and Buplerum falcatum [256]. Specifically, the antidepressant effect of CSS is mediated by increased PI3K/AKT/BDNF signaling and decreased GSK-3β and IL-2 activity [256], suggesting that polyherbal cocktails may be protected from AD development. HQSJDZ, rich in kaempferol and quercetin, had cholinergic, anti-inflammatory, and anti-GSK-3β effects [278,306]. Moreover, a cocktail of C. sinensis, Hypericum perforatum, and Bacopa monnieri produced robust antioxidant effects compared to single-herb treatment [298]. These data suggest that polyherbal treatment may be superior to single-herb therapy.
Due to the well-documented effects of quercetin and kaempferol on Aβ, GSK-3β, PI3K/AKT, and multiple pro-inflammatory molecules, it is possible that both phytochemicals, given their abundance, contribute vastly to the anti-AD effects of several herbs. Such herbs include Ginkgo biloba, Camellia sinensis, Glycyrrhiza uralensis, Cyperus rotundus, and Buplerum falcatum. According to the practice of TCM, it is possible that a multi-herb cocktail containing varying amounts of these herbs could alleviate AD symptoms, as seen with current medications, but it may also halt progression relative to a unique multi-modal mechanism. Multiple studies have suggested that the synergistic effects of polyherbal treatments produce greater anti-AD efficacy compared to single-herb treatment [256,278,298]. Thus, the research and development of future AD drugs should consider the applications of these common herbs in future drug cocktails. On the other hand, since clinical trials featuring Ginkgo biloba extracts have demonstrated controversial results on the progression of AD [296], single-herb treatments may be insufficient to treat AD.
This entry is adapted from the peer-reviewed paper 10.3390/biology12111453
This entry is offline, you can click here to edit this entry!