Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Inhibitors of PARP have been designed so that that they can be useful in the clinic by using their synthetic lethality feature. PARP inhibitors are known as the first synthetic-lethality-inducing agents allowed to be used in the clinic. Although PARP1 inhibitors are primarily used in the clinic to treat cancers with an impaired homologous recombination signaling pathway (especially BRCA1 mutation), they have recently been used as part of an adjunctive therapy in other solid cancers. This was based on the possibility that carriers without a HRD are susceptible to PARPi due to their BRCAness dependency.
- PARPi
- PARPi-resistance
- breast cancer
- BRCA2
- PARP
- BRCA1
1. Resistance to Poly(ADP-ribose) Polymerase Inhibition
Inhibitors of poly (ADP-ribose) polymerase (PARP) have been designed so that that they can be useful in the clinic by using their synthetic lethality feature. PARP inhibitors are known as the first synthetic-lethality-inducing agents allowed to be used in the clinic. Although poly (ADP-ribose) polymerase 1 (PARP1) inhibitors are primarily used in the clinic to treat cancers with an impaired homologous recombination signaling pathway (especially breast Cancer 1 protein (BRCA1) mutation), they have recently been used as part of an adjunctive therapy in other solid cancers. This was based on the possibility that carriers without a homologous-recombination-deficiency (HRD) are susceptible to PARPi due to their BRCAness dependency. Possible mechanisms that may lead to the development of a resistance to PARPi are discussed in detail in the following section. The most common PARPi resistance mechanisms detected in patient tumors include (a) dysregulated molecular signaling, (b) reverse mutations, (c) restoration of replication fork stability, and (d) increased drug efflux (Figure 1).
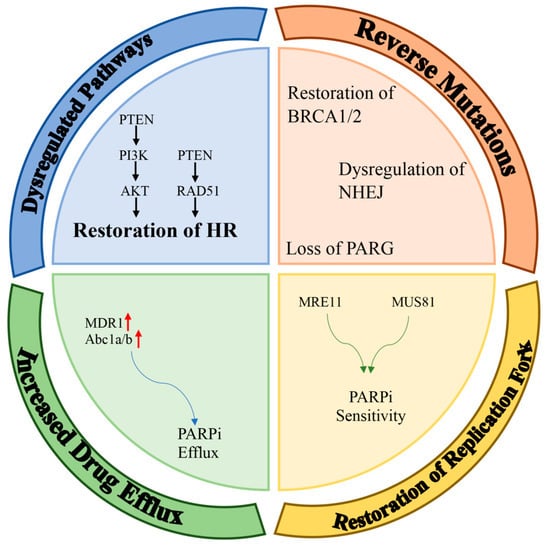
Figure 1. Mechanisms of resistance to PARPi. Potential mechanisms of PARPi-resistance can be classified into four main groups: dysregulation of pathways; restoration of HR; reverse mutations (e.g., restoration of BRCA1/2, dysregulation of NHEJ, loss of PARG); restoration of replication forks; and increased drug efflux (red arrows means up-regulation).
2. Molecular Mechanisms for Resistance to Poly(ADP-ribose) Polymerase Inhibition
Various mechanisms of resistance were identified and are being identified. Understanding these mechanisms is expected to lead to the development of novel strategies to overcome this acquired resistance to PARPi and sensitize BRCA-mutated breast cancer to PARPi in intrinsic resistant cases.
2.1. Dysregulated Molecular Signaling
Phosphatase and tensin homolog (PTEN) is a protein–lipid phosphatase that functions as a tumor suppressor and antagonizes the action of PI3K (Phosphoinositide 3-kinases), resulting in the inactivation of AKT (Protein Kinase B); this consequently inhibits cell growth and cell proliferation [52]. However, AKT activation can also induce BRCA1 expression, which then upregulates AKT’s downstream signaling pathways through a different signaling process [53]. Therefore, irregularities in BRCA levels may affect the PI3K/AKT pathway activation via PP2A (Protein Phosphatase 2). Damage to DSB repair can increase the proliferation of BRCA1-mutant cells via PI3K/AKT activation due to the loss of PTEN [54]. It is known that PTEN contributes to the regulation of RAD51. In fact, low levels of RAD51 have been shown in PTEN knockout mice and PTEN-/- human tumor cells. In addition, a decreased synthesis of nuclear RAD51 have also been shown in PTEN-mutant tumor cells [55]. Interestingly, endometrial cancer cell lines that have lost PTEN have problems with HR DNA repair; therefore, these cells are more sensitive to PARPi [56]. Based on these findings, a loss of PTEN, which is highly associated with BRCA1-mutated breast cancer, appears to promote PARPi-sensitivity. However, it should be noted that the presence of wild-type PTEN also confers a resistance to PARPi in breast cancer [57].
The most frequently activated cellular process in the repair of DNA damage is PARylation, catalyzed by PARP proteins. PARylation is catalyzed by both PARP1 and PARP2. Murai et al. have shown that the siRNA-mediated deletion of PARP1, but not PARP2, abolished olaparib cytotoxicity in prostate cells. Researchers have subsequently shown that PARP1 is the main factor responsible for PARPi-induced cytotoxicity [58]. PARP1 or PARP2 must be attached to DNA for effective PARylation to occur. Preventing PARP from adhering to DNA increases PARPi-resistance, even in the absence of HR. It is clear that the presence of PARP inhibitors, despite the presence of BRCA mutations, will not induce synthetic lethality if PARylation does not occur or if the PARP is prevented from adhering to DNA due to mutations [59].
2.2. Reverse Mutations
The BRCA mutations required for susceptibility to PARPi are reversible and restore protein function. The restauration of these mutations leads to PARPi-resistance. As a result of the restoration of BRCA1/2 function, the frameshift caused by mutation is lost and the open reading frame (ORF) is restored. This causes a full-length wild-type protein to be synthesized in its normal state, resulting in a genetic reversal of the mutation. Lin K.K et al. showed that in BRCA1- and BRCA2-mutated ovarian cancer cell lines, after treatment with cisplatin or PARP inhibitors, a protein in the mutated allele was restored, leading to platinum- and PARPi-resistance in these cells [60]. According to new studies, the return of the mutation found on BRCA1 and BRCA2 may cause resistance to PARPi and limit the success of the PARPi treatment.
Studies demonstrated that a HR-deficiency leads to the activation of the non-homologous end joining (NHEJ) pathway; HR mutations, together with PARP inhibitors, lead to genomic instability and cell death. The decision on whether to use NHEJ or HR to repair DSBs is determined by multiple mechanisms, including cyclin-dependent kinase (CDK) activity and the activation of HR. NHEJ is active during the interphase; meanwhile, HR is active during the S and G2 phases of the cell cycle. In addition, during the S/G2 phases, HR is activated by the binding of the MRE11–RAD50–NBS1 (MRN) complex to the DSB terminals. To ensure synthetic lethality by PARPi in HR-deficient cells, NHEJs need to function properly. When NHEJ is blocked, the inhibition or downregulation of PARP in BRCA1, BRCA2, or ATM-deficient cell lines can escape cell death [61]. Therefore, a partially functional NHEJ mechanism in cancer cells can lead to a loss of PARPi-sensitivity.
PARylation is reversed by PAR glycohydrolase (PARG) via the breakage of PAR chains. Therefore, PARG works in the same framework as PARP inhibitors by preventing the accumulation of PAR chains. A loss of PARG in cell lines with BRCA1 and BRCA2 mutations appears to be a cause of PARPi-resistance. A loss of PARG results in a reduced DNA retention of PARP1 in PARP-inhibitor-treated cells and a partial amelioration of PARP1-induced DNA damage. Overall, the results of these experiments suggest that endogenous PARG activity is required for the cytotoxic effects of PARPi [62].
2.3. Restoration of Replication Fork Stability
BRCA1 and BRCA2 are also responsible for inhibiting the progression of the replication fork after DNA damage [63]. In the absence of BRCA1/2, MRE11 and MUS81 nucleases target the replication fork and cause its collapse and chromosomal aberrations. PTIP and EZH2, following the insertion of MRE11 and MUS81 into the replication fork, are known to cause PARPi-sensitivity [64]. Another factor responsible for the stabilization of the replication fork is RADX. While RADX prevents a MUS81-mediated fork collapse, it also prevents excessive remodeling of the replication fork, as mediated by RAD51 [65]. SMARCAL1, ZRANB3, and HLTF are involved in the modulation of the replication fork. They are required for the degradation of MRE11-dependent nascent DNA in cell lines with BRCA1 and BRCA2 mutations, as PARPi-resistance develops in the absence of SMARCAL1, ZRANB3, and HLTF [66].
Another factor impacting the replication stress is SLFN11; although, recent studies have shown that it does not directly affect the stability of the replication fork; it could rather arrest the replication fork. Replication fork arrest via SLFN11 also causes prolonged replication arrest in the S phase. This prolonged replication fork arrest induces replisome resolution and fork breakage, leading to hypersensitivity to PARPi [67].
2.4. Effect of the Increased Drug Efflux
In addition to the resistance mechanisms specific to the DNA damage response, there are pharmacological factors regulating the response to PARPi. Recent studies suggest that the PARPi response is shaped by ATP binding cassette (ABC) transporters. The increased expression of Multidrug-Resistance Protein 1 (MDR1) [P-glycoprotein (PgP) efflux pump] in tumor cells, which is an important ABC transporter, increases the extracellular excretion of the drugs, resulting in a decrease in the effectiveness of PARP inhibitors [68]. In a BRCA1-mutant mouse model, PARPi-resistance developed when the Abcb1a and Abcb1b genes encoding the PgP pumps were upregulated [69]. Thus, that PARPi-resistance may be associated with an increased expression of drug efflux transporter genes; this resistance is mediated specifically by the Abcb1a/b genes. Rottenberg S. et al. showed that Abcb1a/b expression increased by two-fold up to eighty-five-fold in olaparib-resistant BRCA-mutated breast cancer cells [69]. Similarly, Abc1a/b expression in ovarian cancer cells has been shown to be associated with a resistance to olaparib and rucaparib. The administration of the MDR1 inhibitor tariquidar in ovarian and breast tumors has also been shown to sensitize tumors to PARPi [70,71]. In the meantime, a treatment with one of the Abcb1a/b inhibitors, verapamil or elacridar, was also able to reverse PARP-resistance [71]. However, these approaches have not been shown to reverse PARP-resistance in clinical trials.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15143642
This entry is offline, you can click here to edit this entry!