Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Nutrition & Dietetics
Obesity affects more than one billion people worldwide and often leads to cardiometabolic chronic comorbidities. It induces senescence-related alterations in adipose tissue, and senescence is closely linked to obesity. Fully elucidating the pathways through which vitamin D exerts anti-inflammatory effects may improve our understanding of local adipose tissue inflammation and the pathogenesis of metabolic disorders.
- adipose tissue
- obesity
- senescence
- vitamin D
- inflammation
1. Introduction
Adipose tissue (AT), traditionally considered an inert organ primarily responsible for energy storage and release, has emerged as a subject of increasing research interest in recent decades. Studies have revealed that white adipose tissue (WAT) has far more intricate functions than previously realized, exhibiting numerous connections and being responsible for multiple processes within the body. As one of the largest endocrine organs, WAT actively synthesizes hormones such as adiponectin, adipsin, apelin, leptin, resistin, and visfatin. These hormones are involved in regulating inflammation, insulin secretion, insulin sensitivity, and food intake, and they respond to both environmental and internal cues by releasing proinflammatory cytokines [1].
In addition to adipose cells, the stromal vascular fraction (SVF), which is regarded as the local “immune system”, plays a crucial role in AT. It surrounds crown-like structures and perivascular spaces and comprises mesenchymal, endothelial, and immune cells such as macrophages, eosinophils, and Th2 CD4+ T cells [1]. These cells interact with sympathetic nerve endings and produce neurotrophic factors, impacting local sympathetic activity [2]. Diverse macrophage subpopulations in AT can determine whether the environment is pro- or anti-inflammatory, with M1 macrophages releasing proinflammatory cytokines (TNFα, IL-1β, IL-12, and IL-23) and M2 macrophages producing anti-inflammatory cytokines (IL-10) [3,4]. White-adipose-tissue-resident multipotent stromal cells (WAT-MSCs) have been shown to support both group-2 innate lymphoid cell (ILC2) activity and the proliferation of adipose tissue eosinophils (ATEs); the latter play a crucial role in maintaining the M2 polarization of macrophages [5,6].
In humans and rodent models, obesity is characterized by an imbalance in the growth and size of adipocytes, with AT exhibiting hypertrophy in comparison to the surrounding vascular tissue [7]. This imbalance results in regional hypoxia, apoptosis, chemokine release, and inflammatory cell recruitment. This process initiates local inflammation, later leading to systemic subclinical inflammation [7]. The metabolic disruptions contribute to the ongoing cycle of AT dysfunction and inflammation, resulting in the development of a diversity of systemic problems in patients with obesity.
Ageing leads to a rise in the percentage of body fat and a shift in the distribution of AT from the subcutaneous layers to visceral layers (visceral AT, VAT). AT senescence increases the accumulation of senescent cells and alters the preadipocyte cell phenotype, resulting in elevated secretion of proinflammatory cytokines, which consequently contribute to the inflammageing of VAT.
Although inflammation is common in both obesity and senescence, there are key differences between obesity-related AT inflammation and inflammageing with respect to their causes and mechanism and the types of cells and inflammatory cytokines involved. Obesity is associated with early and pronounced AT senescence, and AT senescence increases the risk for obesity, suggesting that obesity and senescence, while not identical, interact and contribute to the development of subclinical inflammation associated with insulin resistance and metabolic syndrome.
Individuals with both obesity and vitamin D insufficiency exhibit elevated levels of proinflammatory cytokines, such as IL-6 and TNFα (tumor necrosis factor α). An umbrella meta-analysis showed beneficial results for vitamin D supplementation on obesity-associated inflammation [8]. Moreover, treatment with vitamin D suppresses the generation of inflammatory markers induced by the NF-κB (nuclear factor kappa B subunit 1) pathway in human adipocytes and preadipocytes. Furthermore, the molecular mechanism by which vitamin D supplementation impacts NF-κB signaling in hypertrophic and inflamed AT is not fully understood [9]. Of particular significance is the phenomenon of adipose cell senescence, and its link to obesity has recently attracted significant research attention. The NF-κB pathway is just one of the common pathways through which vitamin D may modulate both local inflammation and inflammageing. Identifying the common target cells and pathways of vitamin D in adipose cells and the SVF may be of interest for better understanding the mechanisms of obesity and AT senescence. This avenue of investigation has the potential to yield valuable insights and targets for improving metabolic health.
2. Vitamin D and Inflammation in Obesity
2.1. Adipocyte Dynamics in Obesity: Connections with Cellular Senescence
During the ageing process, damage from various agents accumulates in cells, which triggers senescence, leading to the loss of the cells’ ability to divide and exhibit specific characteristics [10]. Cellular senescence occurs due to various factors: DNA damage, telomere shortening, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, mitotic stress, oxidative stress, and oncogene activation [11,12,13]. DNA damage, especially DNA double-strand breaks, activates a response pathway, culminating in the activation of the p53/p21 axis, thereby inducing cell cycle arrest [13]. This is characterized by irreversible cell cycle arrest in the G1 or G2 phase, orchestrated by tumor suppressor proteins and kinases such as p53, p16, and p21 [1,14,15]. Although senescent cells do not proliferate, they remain metabolically active, displaying alterations in gene expression and chromatin structure [1].
AT is particularly susceptible to ageing, as it is among the most-affected tissues by age-related deterioration [16] and is one of the organs where cellular senescence begins earliest [17]. Life expectancy declines with obesity, and fat mass increases with age in both mice and humans. Caloric restriction increases lifespan, which is attributed to a reduction in the volume of VAT depots [18]. Obesity is associated with age-related changes and an elevated number of senescent cells in AT [19].
Cellular senescence, while advantageous for tissue repair and tumor suppression, can be detrimental when chronic, such as in obesity. One of the primary regulators of lipid homeostasis, SREBP1c (sterol regulatory element-binding protein 1), known to modulate cholesterol and fatty acid metabolism, appears to play a crucial role in protecting against adipocyte senescence. Obesity-induced DNA damage triggers adipocyte senescence. The protective role of SREBP1c involves the maintenance of genome stability, increasing PARP1 (poly (ADP-ribose) polymerase 1)’s DNA repair capability. Adipocytes deficient in SREBP1c show increased inflammation, implying that adipocyte senescence might be an early event preceding inflammatory responses in WAT in the context of obesity [20,21].
Senescence and obesity both result in increased cellular dysfunction and inflammation. However, senescence is characterized by an increase in the number of nondividing, inflammatory cells, while obesity is characterized by expansion and proliferation of adipocytes, which are accompanied by metabolic disruptions (Figure 1).
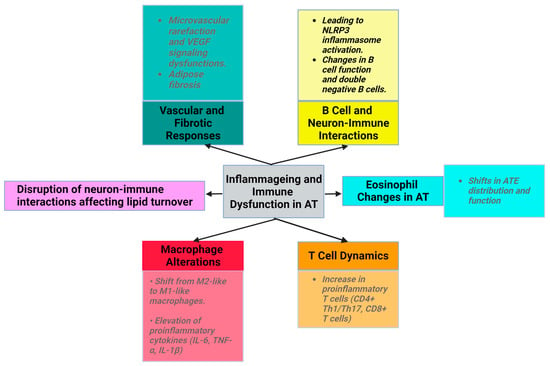
Figure 1. Characteristics of senescence in AT (created with Biorender.com (accessed on 11 December 2023)).
2.1.1. Senescence-Associated Secretory Phenotype (SASP)
A notable characteristic of the senescent cells is their unique secretome, known as the senescence-associated secretory phenotype (SASP). This includes elevated senescence-associated β-galactosidase (SA β-gal) activity, an upregulated expression of cyclin-dependent kinase inhibitor-1 A (CDKNlA) and p53, and an increased production of proinflammatory cytokines and chemokines [22,23]. SASP includes inflammatory and profibrotic factors, which can induce secondary senescence in adjacent cells in a manner modulated by transcription factors, such as NF-κB and C/EBP (CCAAT/enhancer-binding protein) [10]. SA β-gal activity increases in AT as the body mass index (BMI) increases [24]. In patients with obesity, the number of senescent SA β-gal-positive cells is significantly higher in visceral white adipose tissue (vWAT) than in subcutaneous tissue (scWAT).
2.1.2. Preadipocyte Changes in Obesity and Senescent AT
The shift in AT distribution from subcutaneous to visceral depots associated with ageing [1,26] results from the impaired differentiation of mesenchymal progenitors into adipocyte-like cells [2,26].
A series of transcription factors orchestrate adipogenesis and preadipocyte differentiation from pluripotent stem cells under the guidance of specific secretory proteins. Although the preadipocyte count remains stable or even increases with age, there are significant alterations in preadipocyte functionality, characterized by decreased replication and differentiation, increased susceptibility to lipotoxicity, and elevated proinflammatory cytokine production [24]. As these cells age, they secrete markers of the SASP, including activin A, an antiadipogenic factor [27]. The concentration of activin A is strongly associated with age and the occurrence of metabolic syndrome, making it a reliable biomarker for senescence [28].
Elevated activity of SASP markers, particularly p53 and p16, is observed in senescent preadipocytes with impaired differentiation [27]. Ageing and lipotoxicity exacerbate the transformation of preadipocytes into macrophage-like entities, which promotes their proinflammatory effects [1,19].
The number of senescent preadipocytes increases in individuals with obesity, with the number of these cells being up to 30 times higher in people with obesity than in lean individuals [19]. SA β-gal-positive cells are more numerous in cultured preadipocytes obtained from obese animals than in those isolated from lean controls [24].
2.1.3. Neural and Vascular Dysfunction in Obesity and Senescent AT
Ageing disrupts neuron-immune interactions [2], which is related to diminished sympathetic stimulation in elderly individuals. This affects lipid turnover, promotes WAT accumulation, reduces starvation resistance, and potentially regulates body temperature [30]. The exact mechanisms underlying reduced adipocyte lipolysis in ageing remain unclear. Despite normal catecholamine signaling, the diminished adipocyte lipolysis associated with ageing is believed to be governed by AT macrophages through an NLRP3 (NLR family pyrin-domain-containing 3) inflammasome-dependent mechanism [1,31].
Additionally, vasculature dysfunction manifests as microvascular rarefaction due to altered vascular endothelial growth factor (VEGF) signaling in aged mice (Figure 1). However, VEGF-treated mice show an increased lifespan and decreased age-related dysfunction, such as visceral obesity and inflammageing [32]. During physiological ageing, VAT releases profibrotic factors, notably transforming growth factor beta (TGFβ) and osteopontin (OPN). OPN plays roles in cell–matrix interactions across various cell types.
Figure 1. Characteristics of senescence in AT (created with Biorender.com (accessed on 11 December 2023)).
Endothelial cells in AT play a pivotal role in lipid transportation and maintaining WAT stability. Obesity disrupts this equilibrium, causes vascular damage in AT, and induces inflammation [1,34]. VAT-derived microvascular endothelial cells (MVECs) from obese individuals exhibit a more pronounced senescent phenotype than subcutaneous AT-derived MVECs [35].
2.1.4. Low-Grade Inflammation in Obesity and Inflammageing
Ageing is associated with an increase in low-grade inflammation and the dysregulation of the immune function (inflammageing) [37]. Immune cells with proinflammatory properties (CD8+ T cells, Th1, Th17, Tγδ, and B cells) infiltrate the SVF and increase the abundance and size of crown-like structures of AT in animals and humans with obesity [34].
Ageing induces alterations in macrophage activity, accompanied by disrupted phagocytosis and cellular metabolism. The macrophage ratio shifts towards the proinflammatory phenotype, with a decrease in M2 macrophages and CD11c CD206 double-negative macrophages. The levels of proinflammatory cytokines (IL-6, IL-1β, and TNF-α) produced by M1 macrophages are elevated in the AT of older individuals [2]. NLRP3 inflammasome activation in older macrophages elevates proinflammatory cytokine release, contributing to age-related fat accumulation in the bone marrow, impacting hematopoiesis and bone health [1,2,3,40,41].
With ageing, ATEs exhibit shifts in distribution and function. Eotaxin, a pro-ageing factor, accumulates with age, and an increase in its expression is associated with a change in ATE distribution, leading to inflammageing and immunosenescence. Young eosinophils restore WAT function in aged rodents to reduce inflammation, emphasizing eosinophils’ role in AT homeostasis [2,43,44,45].
The number of proinflammatory T cells, such as CD8+ T cells and CD4+ (Th1/Th17), increases with ageing in AT [1]. In aged mice, regulatory T cells (Tregs) maintain AT homeostasis, emphasizing the importance of immune modulation in ageing [7,46]. In animal studies, high-fat diet consumption was found to decrease the levels of the anti-inflammatory cytokine IL-10 in Tregs from VAT. The development, maintenance, and specific gene expression patterns of VAT-Treg cells depend on IL-33 [45,47].
VAT in obese mice is rich in senescent T cells, which are uniquely positive for CD4. Upon stimulation, these cells secrete OPN, a cytokine that has been associated with inflammatory processes in the VAT of individuals diagnosed with obesity and T2DM. Furthermore, patients with obesity and hyperinsulinemia display increased expression of senescence markers in mature adipocytes.
2.1.5. Extracellular Vesicles and Particles (EVPs)
AT secretes EVPs that transport microRNAs (miRs) resistant to degradation, which are important for interorgan communication. EVPs impact metabolic processes and obesity, indicating that AT is a key exosomal source of miRs [50]. In obesity, the expression of miRs (such as miR-27b-3p, miR-122, and miR-192) changes, leading to metabolic imbalances such as insulin resistance [50]. In the context of obesity, the activation of M1 macrophages is stimulated by the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway in conjunction with NF-κB and MAPK (mitogen-activated protein kinase), and miR-155 promotes this process by inhibiting the production of essential anti-inflammatory mediators [51].
EVPs are also linked to cellular ageing, as they release pro-senescence molecules. Furthermore, miRs are essential players in the progression of cellular senescence; the association of miR-155 with telomere shortening underscores their role in ageing [51,52]. Ageing results in an increase in the levels of proinflammatory markers, a phenomenon suppressed by SIRT1, whose levels are modulated by miR-204. Thus, miRs play a crucial role in the complexity and regulation of senescence and may have a significant impact on the relationship between AT senescence and obesity [51,52].
2.2. Physiological Roles of Vitamin D: Bone Health, Immune Function, and Beyond
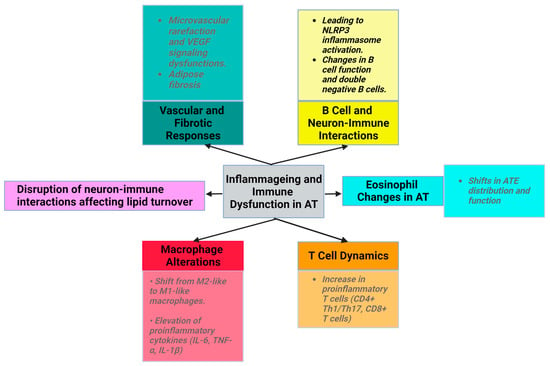
In addition to its critical role in maintaining bone health and regulating phospho-calcium balance, vitamin D exerts an extensive array of effects and is a fundamental regulator of numerous physiological processes. Recent research has revealed that vitamin D regulates the expression of genes associated with the immune system, thereby exerting anti-inflammatory effects. The ability of immune cells (macrophages, B and T cells, and dendritic cells) to produce and express vitamin D receptor (VDR) provides support for this hypothesis [10].
Vitamin D acts through two main mechanisms (Figure 2). First, it acts on its nuclear receptor, VDR, which is found in several tissues. Upon binding to VDR, 1,25(OH)2D3 acts as a transcription factor that regulates the expression of certain genes. These genes can be classified as primary and secondary targets. The primary target genes include transcription factors whose expression is directly influenced by activated VDR following 1,25(OH)2D3 binding. These primary target genes subsequently downregulate the expression of secondary target genes, allowing vitamin D to exert anti-inflammatory effects [10]. Both adipocytes and immune cells express the VDR gene and enzymes associated with the effect of vitamin D. This highlights the critical role of vitamin D in ensuring the proper functioning of AT. VDR knockout mice exhibit reduced AT mass, decreased leptin levels, and increased food intake compared to wild-type mice [54].
Figure 2. Intracellular mechanism of action of vitamin D (created with Biorender.com).
Another mechanism through which vitamin D acts involves the assembly of a heterodimer with the retinoid X receptor (RXR). Vitamin D interacts with a specific DNA sequence known as the vitamin D response element (VDRE) in the promoter region of target genes to induce genomic changes.
Vitamin D also acts through a nongenetic mechanism that is independent of VDR and does not have an impact on gene transcription. Vitamin D binds to the membrane-associated vitamin D receptor, leading to the activation of membrane signaling cascades involving phospholipase C (PLC), diacylglycerol/inositol (DAG/IP3), protein kinase C (PKC), and MAPK, which are critical for translating extracellular signals into appropriate cellular responses [60]. Adipocyte survival, lipid metabolism regulation, insulin response, cell differentiation, inflammatory mediation, and adipokine secretion are all significantly influenced by these signaling cascades. This mechanism is essential for the preservation of cellular function and metabolic homeostasis. These pathways play an essential part in regulating fat storage and breakdown, in addition to influencing the insulin sensitivity of cells, as well as impacting the transition from preadipocytes to mature adipocytes and their involvement in inflammation and overall systemic metabolism.
2.2.1. Vitamin D Deficiency in the Context of Obesity
The optimal level of vitamin D, reflected by the concentration of 25-hydroxyvitamin D (25(OH)D) in the serum, ranges from 30 to 50 ng/mL (75 and 125 nmol/L) [61,62,63]. The Endocrine Society Clinical Practice Guideline defines vitamin D sufficiency as a serum 25(OH)D level of 30–100 ng/mL [64,65]. Vitamin D insufficiency is defined as serum 25(OH)D levels between 20 and 30 ng/mL (50 and 75 nmol/L), and vitamin D deficiency is defined as a serum 25(OH)D level below 20 ng/mL (50 nmol/L) [61].
Vitamin D deficiency is a frequent finding in overweight or obese people. Initially, it was hypothesized that AT functions as the primary reservoir for vitamin D [66]. The trapping hypothesis proposes that fat cells “capture” vitamin D, preventing it from being efficiently metabolized or used by the body. This finding may explain the relationship between obesity and reduced levels of circulating vitamin D, as well as the need for individuals with obesity to take higher dosages of vitamin D to attain comparable blood levels to those with lower adiposity [11,17,24,26,27].
Individuals with obesity have a greater overall level of vitamin D in AT compared to persons with normal weight. However, it was found that the concentrations of vitamin D per gram of fat tissue were the same between individuals with obesity and those with normal weight [67].
2.2.2. The Role of Vitamin D in the Senescence Process
The results of epidemiological surveys in which vitamin D deficiency was found to be associated with a higher mortality rate suggested a role for vitamin D in the process of ageing [77]. Additionally, according to previous research, women with vitamin D deficiency have a reduction in telomere length, which is associated with a comparatively decreased lifespan. A previous study found a significant relationship between serum vitamin D levels and telomere length. This significant correlation persisted even after controlling for factors such as age, physical activity, the use of hormone replacement therapy, and menopausal status. The difference in leukocyte telomere age between subjects in the extreme tertiles of vitamin D concentrations was found to be approximately 5.0 years. This difference was further amplified by increased levels of C-reactive protein (CRP) [78]. Furthermore, mice with VDR deletion display characteristics associated with ageing [79,80].
2.3. Exploring the Shared Characteristics of Obesity and Ageing with a Focus on Vitamin D
Obesity is known to induce a persistent, mild, yet substantial inflammatory response, which has profound implications for overall health, particularly in the context of cardiovascular disease, diabetes, and metabolic syndrome. Cellular senescence is distinguished by the production and release of signaling molecules, including cytokines and chemotactic agents, which collectively reflect the SASP. This process is governed by many factors [52], which also play a role in the subclinical inflammation seen in obesity.
This subclinical inflammation involves intricate cellular pathways, in conjunction with macrophage infiltration and ER stress. These pathways can amplify the activity of one another synergistically, promoting a chronic state of low-grade inflammation. In addition, inflammageing and subclinical inflammation, particularly in the context of obesity, share mechanisms such as the TLR pathways, NF-κB pathway, NLRP3 inflammasome activation, oxidative stress, mitochondrial dysfunction, immune cell population alterations, and gut microbiome changes.
Vitamin D, which is recognized for its immunomodulatory and anti-inflammatory properties, may target specific pathways connected to adipose cell senescence, inflammageing, and subclinical inflammation. These commonalities represent targets for therapeutic interventions for multiple age- and obesity-related health issues.
2.3.1. Toll-like Receptors (TLRs)
TLRs are a family of proteins that play a critical role within the innate immune system. In the context of obesity, TLR2 and TLR4 act as significant mediators that connect excessive food intake and the occurrence of inflammation. Vitamin D exerts regulatory effects on immune cell responses to TLR activation, hence having an influence on the inflammatory response induced by various ligands, including fatty acids [83].
High-fat diet consumption affects the intestinal microbiome composition, increasing gut permeability to lipopolysaccharides (LPSs), which can enter the bloodstream and stimulate TLRs. This condition, also known as “metabolic endotoxaemia”, can induce systemic inflammation in individuals with obesity [84]. Furthermore, saturated fatty acids have the ability to activate TLR4 in AT, hence activating inflammatory signaling pathways [85,86] and contributing to local and systemic inflammation. Obese db/db mice, which exhibit leptin receptor dysfunction, show increased expression of TLR4 messenger RNA (mRNA) in AT. C3H/HeJ mice, which express nonfunctional mutant TLR4, show decreased adiposity and increased insulin signaling upon consumption of a high-fat diet in comparison to control animals. Silencing TLR4 increases insulin resistance and reduces inflammation in murine models of diet-induced obesity.
Data from in vitro studies conducted by Youssef-Elabd et al. [88] and Vitseva et al. [89] showed that the exposure of human SAT explants and adipocytes to saturated fatty acids resulted in the activation of TLR4 signaling, which led to an increase in NF-κB activity. Moreover, the upregulation of TLR2 and TLR4 on macrophages and adipocytes was caused by the activation of these receptors with agonists (Pam3CSK4 (Pam3CysSerLys4) and LPSs, respectively), which increased the production of several inflammatory mediators. These results provide comprehensive evidence of the potential involvement of TLRs in the inflammatory response induced by obesity.
Chronic inflammation is a well-established catalyst of cellular senescence. The persistent activation of TLRs in AT might indirectly facilitate the onset of adipocyte senescence by sustaining the inflammatory response. The activation of TLRs induces the production of ROS and proinflammatory cytokines. The presence of abundant ROS has the potential to cause DNA damage and elicit various forms of cellular stress, both of which may serve as stimuli for the onset of cellular senescence [91].
Given the function of TLRs in obesity-associated inflammation and metabolic dysfunction, they are potential therapeutic targets for obesity and metabolic diseases. Inhibitors of TLR signaling might help to reduce inflammation and improve metabolic outcomes in obesity [94]. Numerous studies have proposed that vitamin D may play a role in obesity by altering TLR activity and thus influencing AT inflammation and senescence [61,95,96,97]. Vitamin D modulates the expression of TLRs on various immune cells.
In summary, while vitamin D can influence TLR activation and has potential implications for AT inflammation and senescence, the detailed mechanisms, especially in the context of obesity, are still under investigation. Considering the multifaceted role of vitamin D in immune modulation and cellular health, the relationship between vitamin D and TLRs remains an important research topic.
2.3.2. NF-κB Pathway
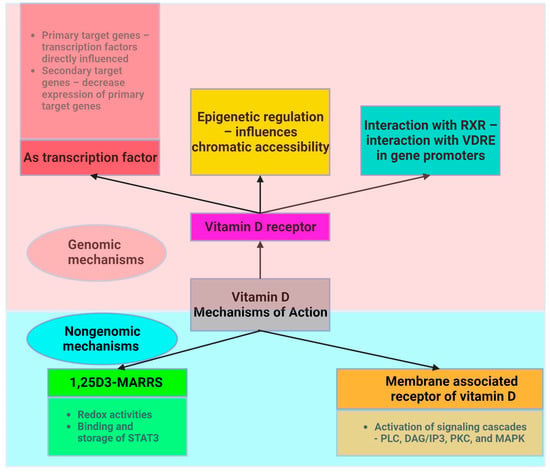
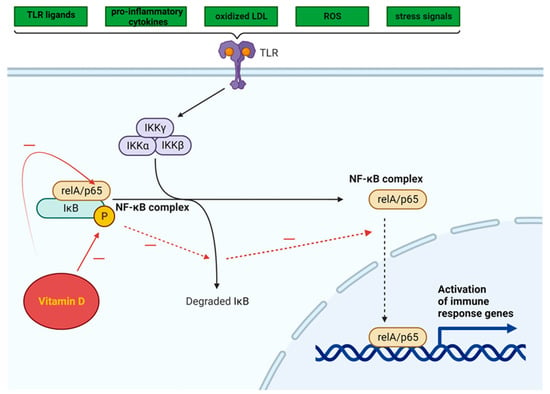
The NF-κB pathway is activated by a variety of TLR ligands and proinflammatory cytokines, oxidized LDL, ROS, and stress signals (Figure 3). The activation of NF-κB leads to the upregulation of miR-34a, which suppresses the production of nicotinamide adenine dinucleotide (NAD) and decreases the levels of sirtuin 1 [101]. In both obesity and ageing, the decrease in the production of sirtuin 1 may involve the NF-κB pathway. The activation of NF-κB and IL-1α in senescent cells leads to a reciprocal induction of these factors, resulting in a positive feedback loop that triggers the synthesis of SASP components [102].
Figure 3. Intracellular NF-κB pathway and influence of vitamin D (adapted from “NF-KB signaling pathway”, by Biorender.com (2023). Retrieved from https://app.biorender.com/biorender-templates, accessed on 12 November 2023).
Vitamin D can inhibit the NF-κB pathway, potentially suppressing the inflammatory response triggered by TLR activation in AT [104]. By impeding the phosphorylation and degradation of the IκB protein (inhibitor of kappa B, a protein that binds the NF-κB transcription factor and maintains NF-κB in an inactive state), the 1,25(OH)2D3-VDR complex hampers NF-κB translocation to the nucleus.
The effect of 1,25(OH)2D3 administration on NF-κB signaling pathways was evaluated in studies using cultured cells, which yielded promising results. An investigation into the impact of high-fat diet consumption on the production of proinflammatory cytokines by stromal vascular cells (SVCs) and adipocytes in lean and obese mice revealed that in vitro treatment with 1,25(OH)2D3 significantly decreased TLR2 expression and increased the mRNA levels of IκBα in SVCs [106].
Vitamin D can suppress the NF-κB signaling pathway by decreasing the expression of miRs (miR-146a, miR-150, and miR-155) in adipocytes derived from both humans and mice. When fed a high-fat diet, miR-146a−/− mice gained more weight and accumulated more fat than wild-type mice, in addition to developing insulin resistance, glucose intolerance, and liver steatosis. This suggests that miR-146a plays a role in maintaining glucose balance both systemically and in adipocytes and may help suppress proinflammatory pathways associated with obesity [108]. The administration of vitamin D to mice fed a high-fat diet resulted in a significant decrease in miR levels [109].
2.3.3. NLRP3 Inflammasome Activation
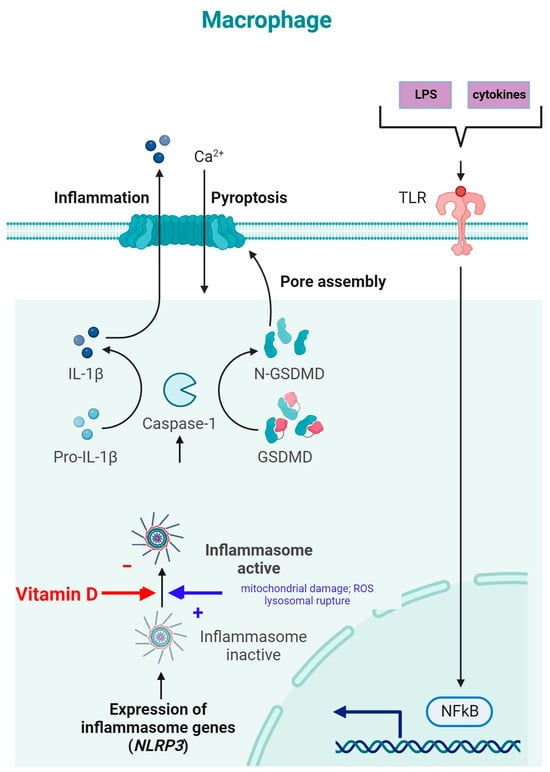
NLRP3 is an intracellular complex composed of multiple proteins that is primarily expressed in macrophages. It is critical for the innate immune response, as it is responsible for identifying and reacting to specific microbial signals, cellular injury, and metabolic abnormalities [40,117]. NLRP3 inflammasome activation requires cell priming, triggered by the binding of microbial components (LPSs) and cytokines (TNF-α) to TLRs. This activates the NF-κB pathway, which induces the transcription and synthesis of NLRP3, which remains inactive. The activation of the NLRP3 inflammasome is caused by numerous factors, such as mitochondrial damage, ROS production, and lysosomal rupture [40]. After NLRP3 activation, the inflammasome is assembled through the activation of caspase-1 and the conversion of IL-1β and IL-18 into their mature, functional forms, which are then released from the cell to promote inflammation and pyroptosis [118] (Figure 4).
Figure 4. NLRP3 inflammasome modulation by vitamin D (adapted from “Suppression of Inflammasome by IRF4 and IRF8 is Critical for T Cell Priming”, by Biorender.com (11 December 2023). Retrieved from https://app.biorender.com/biorender-templates (accessed on 11 December 2023)).
The inhibition of NLRP3 inflammasome activation by vitamin D may reduce the secretion of IL-1 and IL-18. Vitamin D acts as a regulator of NLRP3 inflammasome activation by modulating the activity of macrophages and dendritic cells. In a study on mice with obesity and asthma, vitamin D3 levels were related to the increased expression of IL-1β mRNA and NLRP3 mRNA in lung tissue [119].
Vitamin D can influence cellular ROS levels, and this could be one of the mechanisms through which it modulates NLRP3 inflammasome activity [120]. ROS and cellular oxidative stress may be induced by vitamin D metabolites due to increased lipid oxidation; therefore, 1,25(OH)2D3 was postulated to be a “priming” molecule that can indirectly induce NLRP3 inflammasome activation to promote IL-1β maturation [120].
2.3.4. Immune Cell Modulation
Inflammation can be reduced by vitamin D, which influences the activity of dendritic cells, macrophages, and T cells and affects both the innate and adaptive immune systems [12]. 1,25(OH)2D3 is essential for the downregulation of numerous cytokines in preadipocytes and adipocytes, including MCP-1, IL-1β, IL-6, and IL-8 [106,121,122]. The presence of VDR in both adipocytes and macrophages suggests the crucial roles of vitamin D and its metabolites in inflammatory processes within the AT [123,124].
The Anti-Inflammatory Action of Vitamin D in Cultured Cells
The anti-inflammatory properties of vitamin D were demonstrated in research involving the incubation of adipocytes with 1,25(OH)2D3 for either 24 or 48 h before inflammatory stimuli were introduced [57,66,114,115]. In response to 1,25(OH)2D3, the mRNA levels of proinflammatory cytokines (IL-6, IL-8, and CD14) decreased in human adipocytes and 3T3-L1 preadipocytes [121,122]. The possible mechanisms by which vitamin D exerts its anti-inflammatory effect include a decreased protein expression of TLR-2 and TLR-4, an increased mRNA expression of trans-acting T-cell-specific transcription factor (GATA-3) via the upregulation of the upstream factor signal transducer and the activator of transcription 6 (STAT6), decreased levels of pp38 (phosphoprotein 38) and p42/44 (ERK1/2), and an altered localization of p65 [125].
In an observational study on vitamin-D-deficient women with obesity, macrophage infiltration of AT was dose-dependently associated with serum vitamin D levels. Participants with moderate vitamin D deficiency exhibited a greater concentration of proinflammatory cytokines than those with mild deficiency [126]. Supplementation with vitamin D3 decreased the production of proinflammatory cytokines: TNFα, PAI-1 (plasminogen activator inhibitor-1), IL-6, and ROS and pro-fibrotic genes in studies on AT biopsies from patients with obesity and 25(OH)D insufficiency [127].
Inconsistent Results in Human Studies
Research examining the effect of vitamin D supplementation on serum levels of inflammatory cytokines in humans has yielded inconclusive findings [131,132,133]. Vitamin D supplementation increases the levels of anti-inflammatory cytokines (IFN-γ and IL-10) in subjects with normal weight and vitamin D insufficiency [134]. While some studies have suggested that increased serum 1,25(OH)2D3 concentrations are associated with decreased inflammatory marker expression in individuals with normal weight [135], other research has found no significant effects of vitamin D on inflammatory biomarkers in patients with obesity [136,137,138,139,140,141,142,143]. Furthermore, studies on the effect of vitamin D supplementation on CRP serum levels in patients with obesity have yielded conflicting results [137,138,139,141,142,143,144,145,146,147,148,149,150,151,152,153,154]. A key factor contributing to this inconsistency is the variation in study design across different trials. First, the sample population of the RCTs ranged from individuals with normal weight to those with obesity, as well as from patients with T2DM and those without T2DM.
Insights from Meta-Analyses
A meta-analysis conducted by Yu et al. [155] showed that vitamin D supplementation reduced CRP levels but did not impact IL-6 or TNF-α levels. The analyzed studies varied in terms of design, vitamin D dose, and duration. Another meta-analysis by Jamka et al. [156] did not find a significant effect of vitamin D supplementation on inflammatory marker levels in overweight and obese patients. Few studies were included in this analysis, and they used different vitamin D doses and treatment durations, making it challenging to identify clear patterns of changes [8].
2.3.5. Oxidative Stress
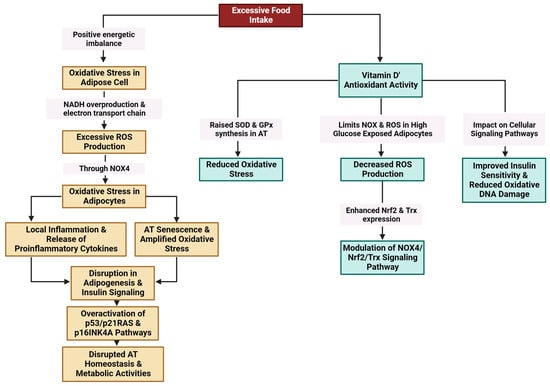
Excessive food intake results in a positive energy imbalance, which favors oxidative stress in adipose cells through NADH (nicotinamide adenine dinucleotide hydrogenase) overproduction and the electron transport chain in mitochondria. Excessive ROS production through NADPH oxidase 4 (NOX4) is linked to oxidative stress in adipocytes [157,158,159,160]. Oxidative stress amplifies local inflammation and is accompanied by the release of proinflammatory cytokines [161]. Exacerbated oxidative stress and local hypoxia due to the imbalance between the supply of and demand for hypertrophic adipose cells together with the release of proinflammatory molecules provoke adipocyte damage [159] (Figure 5).
Figure 5. Modulation of oxidative stress in AT by vitamin D (created with Biorender.com (accessed on 11 December 2023)).
This entry is adapted from the peer-reviewed paper 10.3390/metabo14010004
This entry is offline, you can click here to edit this entry!