1.Mitochondrial Structure and Function in MAFLD
1.1 Mitochondrial Membrane Structure
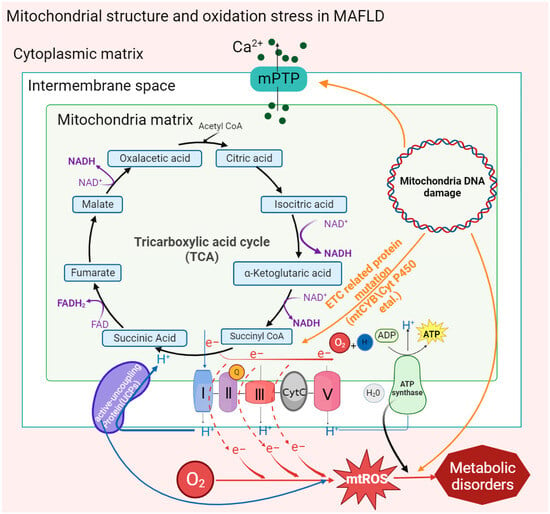
The double-membrane structure of mitochondria provides a place for a variety of metabolic reactions. Porins distributed in the outer membrane are screening sites for metabolic substrates in the cytoplasm, and ion channels or binding proteins distributed in the inner membrane, including respiratory chain proteins, ATP synthases, and vitamin-binding receptors, are highly involved in mitochondrial metabolism. Recently, it has been found in clinical patients and mouse models of MAFLD that abnormal activation of a special type of nonspecific ion channel in the mitochondrial membrane structure, the mitochondrial permeability transition pore (MPTP), is associated with oxidative stress and the development of MAFLD [
32,
41,
42,
43].
In the MAFLD mouse model, it was found that intracellular accumulation of free fatty acids (FFAs) could directly stimulate MPTPs to maintain an open state, and mitochondrial Ca
2+ outflow stimulated related inner membrane proteins, such as adenine nucleotide translocator or F1F0-ATPase, to aggregate, forming new MPTPs [
32]. Moreover, this result was also found in MAFLD patients whose MPTPs could also be activated by mt-ROS and other oxidation byproducts [
42,
44].
Under mitochondrial stress conditions, MPTPs open in inner mitochondrial membranes. Increased membrane permeability leads to Ca
2+ escape from mitochondrial calcium stores and electron transport chain (ETC) protons into the cytoplasm, resulting in typical MAFLD symptoms such as defective ATP production and increased cytoplasmic concentration of Ca
2+ [
45]. Moreover, significant mitochondrial swelling was found in MAFLD mice, including loss of the mitochondrial outer membrane, disappearance of cristae, and expansion of the mitochondrial matrix. These structural changes may also be caused by the abnormal activation of MPTPs through the action of FFAs [
32]. MPTPs are widely involved in cell apoptosis and damage clearance mechanisms, which are related to an increase in membrane permeability leading to the release of cytochrome C into the cytoplasm. In addition, the activation of MPTP reduces the activity of mitochondrial inner membrane proteins and inhibits respiratory chain function and ATP synthesis, thus causing mitochondrial oxidative stress. Reduced oxidation activity of ETC-related complexes Ⅰ and III, cytochrome C, and coenzyme Q on the inner mitochondrial membrane leads to electron leakage, which then combines with oxygen molecules to form mt-ROS [
46]. In MASH fibroblasts, mt-ROS expression and mitochondrial membrane permeability were found to be significantly increased and eventually escaped to other organelles [
43].
A variety of functional proteins are located in the inner mitochondrial membrane. For example, abnormal activation of uncoupling proteins (UCPs) on the inner mitochondrial membrane could significantly reduce the mitochondrial membrane potential and H
2O
2 release, which are significant for the promotion of fat oxidation metabolism [
30]. It is worth noting that decreased membrane potential led more free fatty acids to enter the mitochondria by decreasing the activity of inner proteins in oxidative phosphorylation (OXPHOS) and β-oxidation, resulting in lipotoxic accumulation to promote the deterioration of MAFLD toward MASH [
47].
1.2 Mitochondrial DNA Mutation
In hepatocytes, more than 90% of mitochondrial proteins are encoded by nuclear DNA, while mitochondrial DNA (mt-DNA) encodes the remaining mitochondrial proteins. The expression of mt-DNA directly affects the efficiency of cellular oxidation. According to the genome comparison of clinical patients, the liver mitochondrial circular gene (mt-DNA) of MAFLD patients has a higher gene mutation rate and heterogeneity [
31].
Mitochondrial cytochrome B, encoded by mt-DNA, is an essential component of complex III of the electron transport chain, which is responsible for electron transport and assisting in the formation of the proton gradient. In patients with MAFLD, it was found that the mt-CYB gene was mutated abnormally. The abnormal mt-CYB gene leads to ETC disorder by changing protein activity, stimulating the production and release of mt-ROS and carcinogenic metabolic byproducts (such as 2-hydroxyglutarate), and then aggravating the degree of MAFLD [
48]. Existing studies have shown that increased mutations in mt-DNA and mt-DNA integrity may be associated with oxidative damage in vivo. ROS and peroxyl free radicals oxidize guanosine to a DNA oxidation complex (8-OHdG) and lipid peroxyl free radicals (4-HNE), respectively, to destroy the mt-DNA structure. Then, the damaged mt-DNA reduces ETC activity, which downregulates the OXPHOS response and participates in the pathogenesis of MASH [
49].
In addition, the accumulation of mt-DNA gene mutations in patients with MAFLD was found to directly regulate mitochondrial oxidation reactions, including those encoding carrier proteins localized in the electron transport chain, peroxisome proliferator-activated receptor-γ coactivation factor 1 (PGC-1) and cytochrome P450 in OXPHOS [
50]. The transcription of PGC-1 is involved in the mitochondrial antioxidant mechanism, and the accumulation of cytochrome P450 2E1 (CYP2E1) mutations affects ETC reaction activity and stimulates the accumulation of mt-ROS, resulting in an elevated trend in MAFLD [
51]. In addition, it has also been shown that the induction of elevated CYP2E1 in MAFLD may be an adaptive mechanism to inhibit lipid accumulation and that CYP2E1 metabolizes FFAs in MAFLD through ω-hydroxylation. However, the hydroxylated fatty acids generated during this process are converted into cytotoxic dicarboxylic acids, which contribute to the exacerbation of MASH [
52] (
Figure 1).
Figure 1. Oxidative stress caused by mitochondrial structures and mt-DNA mutations in MAFLD. Activation of the mitochondrial membrane permeability transition pore (MPTP) by factors such as mitochondrial mutant genes and the accumulation of fatty acids promotes the outflow of Ca2+ from the mitochondrial calcium pool and then stimulates the activity of inner membrane proteins to affect the ATP synthesis rate and form new transition pores. Mitochondrial gene mutation (mt-DNA) also stimulates the activation of MPTP and uncoupling proteins (UCPs). In addition, the reduction in the activity of inner membrane proteins leads to electron leakage in the ETC, which promotes the generation of mt-ROS and ultimately aggravates the degree of mitochondrial oxidative stress in MAFLD.
1.3 Mitochondrial Quality Control
Mitochondria are highly dynamic organelles. The imbalance between mitochondrial dynamics is key to determining the total mass of mitochondrial cells in the pathogenesis of MAFLD or MASH models [
53,
54,
55,
56,
57,
58,
59]. Abnormal expression of mitochondrial fusion proteins or giant mitochondria has been found in various mouse models of nonalcoholic fatty liver disease. Yamada, T. et al. found that excessive mitochondrial fusion or giant mitochondrial structure exists in mouse hepatocytes by treating the MASH mouse model fed a high-fat diet (MCD), although targeting Opa1 expression could effectively reduce the formation of giant mitochondria and restore the reduced succinate dehydrogenase (SDH) activity induced by MASH [
60]. Du, J. et al. reported that mitochondria of hepatocytes in vivo were significantly swollen and cristae disappeared in MASH mice induced with a high-fat-high-carbohydrate-high-cholesterol diet (HFHCHCHFHCD). In the MASH model of AML-12 and HepG2 cells treated with palmitic acid (PA), the expression of mitochondrial fusion protein 1 (Mfn1) was significantly downregulated [
61]. In addition, the lack of Mfn2 leads to a reduction in phosphatidyl serine (PS) transfer from the ER to mitochondria, thereby inducing ER stress and lipid accumulation. Hernandez-Alvarez, M.I. et al. found that the expression of Mfn2 significantly decreased in MAFLD patients, and in the MASH mouse model, upregulating Mfn2 alleviated the MASH phenotype [
62].
In the pathogenesis of MAFLD, the dysregulation of mitochondrial dynamics is manifested by increased mitochondrial fission and decreased mitochondrial fusion. The occurrence of liver fibrosis or fat accumulation in hepatocytes and HSCs as very low-density lipoprotein (VLDL) is an important symptom of MAFLD and is regulated by mitochondrial quality control and mitophagy mechanisms. Increased mitochondrial fission by overexpressing Fis1 activated HSCs, and decreased mitochondrial fission by Mdivi-1 treatment induced HSC apoptosis both in vivo and in vitro [
63]. In a mouse liver fibrosis model infected with cercariae of Schistosoma japonicum, Drp1 phosphorylation at Ser637 regulated mitochondrial fission, and decreases in Opa1 and Mfn1 inhibited mitochondrial fusion and resulted in vacuolated structures [
64]. Takeichi, Y. et al. found that mitochondrial fission protein Mff knockout mice were more prone to have MASH phenotypes than normal diet groups when fed a high-fat diet (HFD) [
65]. Moreover, hepatocytes in Mff knockout mice had swollen balloons, and giant mitochondria increased; the expression of mitochondrial peroxisome proliferator-activated receptor-α (PPAR-α) and FAO-related protein (CPT1A) decreased significantly, and the catalyzed oxidative stress response accompanied by inflammatory cell infiltration caused upregulation of fibroblast growth factor (FGF21), which promoted the formation of MASH phenotypes.
Generally, damaged mitochondria are separated from normal mitochondria by initiating fission [
33]. However, mitochondrial fission is affected by stress, and fragmentation accelerates mt-DNA fragmentation, thus stimulating the production of mt-ROS, causing oxidative stress and promoting the development of MAFLD. In addition, related studies have confirmed that Drp1-mediated mitochondrial fission is accompanied by increased levels of proinflammatory factors such as TGF-β, TNF-α, and IL-6 in MASH models. In addition, abnormal mitochondrial fission also affects autophagy pathways [
63,
66]. In the hepatocytes of HFD-fed Mff-KO mice, it was found that p62 and pyruvate dehydrogenase in the mitochondria increased, which inhibited the clearance of damaged mitochondria [
65].
1.4 Mitophagy
Mitophagy is beneficial for removing damaged mitochondria and oxidative toxic byproducts (mt-ROS, dicarboxylic acid, etc.) and maintaining the normal physiological activities of mitochondria in cells.
In recent years, it has been found that mitophagy dysfunction has a nonnegligible regulatory impact on the development of MAFLD. Li, X. et al. found that the abundance of PINK and Parkin in HFD-fed mice was significantly lower than that in the control group, but the expression of P62 and LC3-I/II was significantly increased [
67]. In addition, the expression of PINK1 and Parkin was significantly reduced in CCL4-induced liver fibrosis mice and Kupffer cell (KC)-transformed HSC cell models [
68]. Dou, S.D. et al. reported that the mitochondrial antioxidant ubiquinone (Mito Q) alleviated the decrease in Parkin protein and restored the level of mitophagy during HSC activation [
69]. In the MAFLD mouse model and PA-induced AML-12 cells, the mitophagy pathway mediated by PINK/Parkin/P62 was inhibited. Meanwhile, it was also reported that the decreased activity of peroxisome-activated receptor-γ (NR1C3) inhibited the decomposition of H
2O
2, induced mt-ROS-mediated cellular oxidative stress by inhibiting the PINK/Parkin pathway, and had a negative impact on the clearance of damaged mitochondria [
67].
In addition, in mt-ROS-overloaded mitochondria, cytochrome c is released into the cytoplasm due to decreased ETC activity, causing apoptosis and aggravating oxidative stress. Overexpression of TIM4 upregulated mitochondrial AKT1 in KCs [
68]. In addition, AKT1 could activate PTEN-induced putative kinase 3 (PINK3) to aggravate the mt-ROS-mediated BAD family (BCL2, etc.), which could bind to damaged mitochondria and induce mitophagy. Zhou, T. et al. showed that upregulation of Mst1 in primary mouse hepatocytes treated with PA could activate the expression of phosphorylated AMPK and eliminate the accumulation of damaged mitochondria caused by decreased Parkin-related mitophagy protein activity [
70]. AMPK could target and inhibit the mTOR complex and activate phosphorylated ULK1, thereby eliminating the inhibitory effect of mTOR on ULK1 activation, realizing the recruitment of P62 and migrating to mitochondria, and participating in mitophagy [
71,
72,
73].
Excessive mitophagy causes the release of mitochondrial damage-associated molecules (mt-DAMPs). Mt-DAMPs can be released from damaged mitochondria to the cytoplasm, thereby causing systemic inflammatory response syndrome to promote liver fibrosis [
21,
63,
74,
75]. In the liver, mt-DAMPs are mainly damaged by linking with TLR9 and formyl peptide receptor 1 to activate polymorphonuclear cells to promote MASH inflammation and activate HSCs, causing fibrotic scars [
32,
42]. However, the inhibition of mitophagy also leads to the accumulation of mt-ROS, which leads to hepatocyte necrosis and the massive release of mt-DAMPs [
67]. In addition, the release of inflammatory factors and inflammasomes (NLPR3) accompanying the apoptosis of hepatocytes stimulates immune cell infiltration and HSC activation and promotes the progression of MASH [
15]. NLPR3 is a multiprotein immune complex that is activated by pathogen-associated and danger-associated molecular patterns (PAMPs and DAMPs), such as lipopolysaccharide (LPS) and cholesterol (CHO) [
76] (
Figure 2).
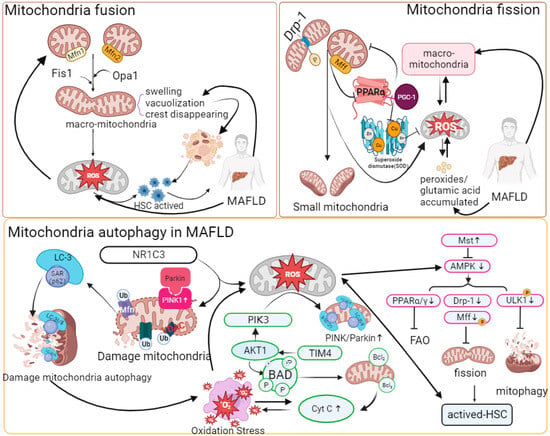
Figure 2. Mitochondrial quality control and oxidative stress in MAFLD.
“↑” represents upregulated expression; “┬” represents downregulated expression. With the stimulation of massive fat accumulation, Mfn1/2, Fis1, or Opa1 has regulatory significance for the formation of giant mitochondria. Changes such as swelling structure and vacuolization of the giant mitochondria result in the accumulation of ROS in mitochondria and HSC activation. The increased expression of phosphorylated Drp-1 and abnormal expression of Mff protein promote mitochondrial fission. A reduction in Mff inhibits the combination of PPAR-α and PGC-1, thereby reducing the synthesis and accumulation of SOD and mt-ROS. Mt-ROS can increase α-SMA, COLLAL-1, TGF-β, TNF-α, and IL-6 to aggravate inflammation and fibrosis. The combination of Parkin and PINK1 promotes the ubiquitination of mitochondrial membrane proteins, and then P62-LC-3 can target and recognize ubiquitinated membrane proteins to initiate autophagy, while downregulation of NR1C3 inhibits the Parkin/PINK1 pathway in MAFLD. At the same time, in HSCs, the increase in related immune proteins, such as TIM4, induces AKT-1 and PIK3 and the release of mt-ROS. Then, mt-ROS stimulate the BAD family and promote the release of Cyt C, causing mitochondrial oxidative stress and autophagy. PIK3 activates Parkin/PINK1. In addition, pathways such as AMPK can compensate for the abnormal mitophagy caused by the inactivation of Parkin/PINK1. AMPK controls HSC activation and mitophagy by activating phosphorylated ULK1, PPAR, and Drp-1. Upregulation of Mst1 expression can activate AMPK in MAFLD. The abbreviations in Figure 2 are defined as follows: Mfn1/2 (Mitofusion 1/2); Fis1 (Fission 1); Opa1 (optic atrophy 1); Drp-1 (dynamin-related protein 1); Mff (mitochondrial fission factor); PGC-1 (peroxisome proliferator-activated receptor gamma coactivator 1); PPAR-α/γ (peroxisome proliferator-activated receptor alpha/gamma); α-SMA (alpha-smooth muscle actin); NR1C3 (nuclear receptor subfamily 1 group C member 3); TIM4 (T-cell immunoglobulin and mucin domain-containing 4); PIK3 (phosphatidylinositol 3-kinase); AKT1 (AKT serine/threonine kinase 1); BAD (BCL2-associated agonist of cell death); Cyt C (cytochrome c); Mst (mammalian sterile 20-like kinase); AMPK (AMP-activated protein kinase); FAO (fatty acid oxidation); and ULK1 (unc-51 like autophagy activating kinase).
1.5 Oxidative Phosphorylation
Oxidative phosphorylation, as an important source of ATP in organisms, is the main metabolic mode of nutrient metabolism. Most of the electrons migrate down through the ETC to cytochrome c oxidase and then combine with protons and O
2− to form water, but there are still electrons leaking from the ETC that directly react with oxygen to form mt-ROS, such as superoxide anion (O
2−), H
2O
2, etc. In recent years, mitochondrial dysfunction and abnormal production of mitochondrial ROS have been widely detected in MAFLD and MASH [
77,
78,
79,
80,
81,
82]. In MAFLD, activation of MPTPs and mt-DNA mutations could reduce the activity of complexes on the ETC, which is the main reason for electron leakage. Mitochondrial gene mutations, quality control, and mitophagy disorders are also important reasons for mt-ROS generation.
To maintain the balance of the mitochondrial oxidation reaction, mitochondria have antioxidation enzymes and antioxidant molecules (glutathione, VE, etc.) to eliminate mt-ROS accumulation by capturing and removing free radicals and harmful substances to repair oxidative damage [
83]. Recently, it has been found that the nuclear factor-κB (NF-κB) pathway has bidirectional effects in different stages of the oxidative stress response in the pathogenesis of MAFLD [
84]. In the early stage of oxidative stress, NF-κB inhibits the continuous accumulation of superoxide and accelerates the clearance of damaged hepatocytes by activating the activity of manganese superoxide dismutase (MnSOD) and the Nf-κB/JNK pathway [
85]. The antioxidant mechanism of mitochondria is limited to dealing with a large number of oxidation products in the inflammatory environment because it was found that inhibitors of NF-κB kinase (IκB kinase, IKK) could be activated by phosphorylation of proinflammatory signaling molecules such as lipopolysaccharide (LPS), tumor necrosis factor (TNF), and interleukin-1 (IL-1) to inhibit the NF-κB pathway [
12,
86,
87], accelerating the release of proinflammatory factors such as TNF-α and IL-1β, positively regulating the accumulation of mt-ROS, and eventually causing the deterioration of chronic inflammation [
88,
89,
90]. JNK is a member of the mitogen-activated protein (MAP) kinases that participate in regulating mitophagy. In addition, the antioxidant factor PPAR coactivator-1 (PGC-1) transcription factor also has a positive effect on the recovery of mitochondrial oxidative stress [
91]. In MAFLD mouse models, upregulated PGC-1α/fibroblast growth factor 21 (FGF-21) expression could restore the copy number of mt-DNA and OXPHOS activity [
92]. However, in the pathogenesis of MAFLD and MASH, it was found that PGC 1α deficiency could directly downregulate superoxide dismutase (SOD), which reduced the activity of molecules such as catalase and glutathione peroxidase and further led to an imbalance of the antioxidant system [
93].
The excessive accumulation of mt-ROS damages mt-DNA and combines with FFAs to participate in lipid peroxidation reactions. Moreover, mt-ROS in hepatocytes can mediate the release of TNF [
16]. TNF leads to MPTP activation, ETC uncoupling, and proton pumping. In MASH mouse models and clinical cases, it was found that the byproducts formed from insufficient oxidation of overloaded FFAs, such as dicarboxylic acid, could bind with uncoupling proteins on the mitochondrial membrane, prompting protons to directly enter the matrix through uncoupling protein channels without interacting with ATP synthase, ultimately leading to a decreased ATP synthesis rate and insufficient energy production [
94]. It has been found that OXPHOS driven by fatty acid β-oxidation in M2 macrophages is converted to the glycolytic pathway in the activated HSC state of mouse models of MAFLD [
95]. The depletion of ATP and the increase in glycolysis activity both maintain the energy demand of cells. However, excessive glycolysis would also lead to an increase in intracellular pyruvate content and TNF or LPS, which has a partial negative effect on the lipid metabolism process of mitochondria and HSC activation.
1.6 Fatty Acid Oxidation (FAO)
Fatty acid oxidation is one of the main oxidation reactions that inhibits lipid accumulation in hepatocytes and provides ATP. Free fatty acids (FFAs) are substrates of various lipotoxic products, such as ceramides and diglycerides, in vivo, which induce metabolic stress and cell death [
12,
96]. FFAs initially decompose fatty acyl-CoA through the mitochondrial outer membrane porin and inner membrane transport protein and then participate in the tricarboxylic acid cycle reaction (TCA) to release electrons and form final products such as water to complete the consumption of liver lipids [
97,
98,
99]. FFAs undergo esterification to form triglycerides that protect hepatocytes from lipo-toxicity at the same time.
The accumulation of lipo-toxicity induced by steatosis and excessive accumulation of FFAs are the hallmark symptoms of the development of MAFLD. Fatty acids can be catabolized in microsomes, peroxisomes, and mitochondria. In recent years, it has been found in clinical patients and animal models of MASH that the expression changes in the PPAR involved in mitochondrial fatty acid β-oxidation and regulated by mt-ROS are significantly correlated with the occurrence of MASH. Sven Francque. et al. found that in MASH patients, the expression of PPARα was significantly downregulated, and in MASH mice fed a diet lacking methionine choline (MCDD), a decrease in PPARα led to an aggravated degree of hepatitis [
100]. Chen, Y. et al. found that PGC-1 was a coactivator of PPARα, and its expression was reduced with the inhibition of PPARα [
101,
102]. In addition, the fatty acid β-oxidation-related proteins CPT1, CPT2, ACOX1, and ACOX2 were also affected and downregulated by the downregulation of PPARα, resulting in the accumulation of lipids in cells and aggravation of the degree of hepatitis. The reduction in CPT1, CPT2, ACOX1, or ACOX2 directly inhibits the permeability of the mitochondrial membrane, preventing FFAs from entering the mitochondria for consumption and metabolism [
103]. In the pathogenesis of MASH, studies have found that a lack of receptor-interacting protein kinase 3 (RIPK3) leads to the upregulation of peroxisome proliferator-activated receptor-γ (PPAR-γ) and fibroblast growth factor 21 (FGF-21) to realize the reverse recovery of MASH [
47].
The other two proteins of the PPAR family, PPAR-β and PPAR-δ, are also widely expressed in hepatocytes, KCs and HSCs. In the different mouse models of MAFLD, activation of PPAR-α/β/δ induces catalase (CAT), promotes the conversion of FFAs to triglycerides to alleviate lipid toxicity, inhibits the generation of mt-ROS and the secretion of IL-1 and TNF in the lipid peroxidation reaction caused by excessive fat accumulation in mitochondria, and finally reduces the occurrence of liver fibrosis and MASH [
104,
105,
106].
In addition, NADH and FADH2 produced by mitochondrial β-oxidation participate in ETC activities by transferring electrons. However, affected by limiting factors such as the ATP synthesis rate, electron transfer in the ETC is prone to leakage, which leads to the accumulation of mt-ROS. mt-ROS inhibit the β-oxidation of hepatic FFAs by reducing the de-palmitoylation activity of the mitochondrial outer membrane protein FAT/CD36, resulting in the accumulation of FFAs in the cytoplasm [
107].
MAFLD is a polygenic disease, and the influence of gene regulation on fat oxidation metabolism is also an important factor that promotes FAO process imbalance in the development of MAFLD. For example, the nonsynonymous mutation of SOD2-C47T can reduce the activity of MnSOD and inhibit the clearance of peroxidation byproducts produced in the process of OXPHOS [
108]; the mutation of UCP3-55T leads to abnormal lipolysis, causing fat accumulation and inducing inflammation [
109]; GCL downregulates GCL, the rate-limiting enzyme for glutathione (GSH) synthesis, and inhibits the clearance of cellular peroxidation, resulting in the accumulation of ROS [
110].
In MAFLD, it was found that in addition to the dysfunction of mitochondrial metabolism in the process of fat accumulation, organelles such as the endoplasmic reticulum (ER) also mediate the exchange of metabolites through polymeric protein complex structures such as mitochondria-associated endoplasmic reticulum membrane protein (MAM) [
29,
111,
112,
113]. When the homeostasis of the ER is imbalanced or there is a lack of energy, the ER is induced by activating the unfolded protein response (UPR), resulting in reduced GSH. The distribution of imbalance between GSH and oxidized glutathione (GSSH) causes mitochondrial stress and realizes the negative regulation of mt-ROS [
21].
In brief, fatty acid β-oxidation serves as another bioenergetic pathway that occurs in mitochondria in addition to oxidative phosphorylation. Abnormalities in β-oxidation could cause the accumulation of intracellular FFAs, aggravate lipo-toxicity, and eventually induce inflammation and steatosis. In MASH patients, the accumulation of mt-ROS plays a negative role in the mitochondrial oxidation reaction and abnormal FAO, finally aggravating the accumulation of lipid substances and the formation of toxic byproducts [
114] (
Figure 3).
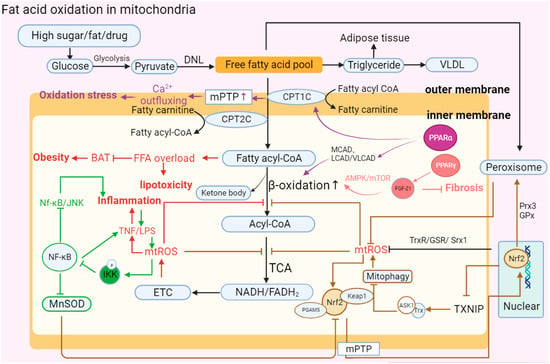
Figure 3. Abnormal fatty acid oxidation in mitochondria. “↑” represents upregulated expression; “┬” represents downregulated expression. Carbohydrates in hepatocytes are initially metabolized into pyruvate, and then pyruvate forms free fatty acids (FFAs) through DNL. Finally, FFAs form triglycerides and VLDL in MAFLD. Upon stimulation such as the introduction of high sugar and fat, the activation of MPTP and decrease in PPARα leads to the abnormal structure of CPT1C and CPT2C, which transport FFAs on the mitochondrial membrane. FFAs flow into the mitochondrial matrix, causing lipotoxic accumulation and further affecting BAT consumption, which leads to obesity. FFAs in the matrix are decomposed into acyl-CoA via β-oxidation and then participate in oxidative phosphorylation. In MAFLD, PPARα can reduce the activity of β-oxidase and inhibit the decomposition of FFAs, while increasing the activity of PPAR-γ can promote the expression of FGF-21 and restore the oxidation of FFAs through the AMPK-mTOR pathway to inhibit the development of fibrosis. Moreover, mitochondrial oxidation imbalance causes the accumulation of mt-ROS, TNF, and LPS, aggravating the development of inflammation. At the same time, TNF can activate phosphorylated IKK to inhibit the NK-κB antioxidant response, including promoting the continuous accumulation of superoxide by reducing MnSOD activity and the Nf-κB/JNK reaction. In addition, Nrf2 mediates oxidation reaction efficiency and participates in fat metabolism. Nrf2 forms a ternary complex with Keap1 and PGAM5 to directly respond to mt-ROS and stimulates nuclear Nrf2 to be accelerated and transported into mitochondria. Nrf2 can eliminate excess mt-ROS accumulation and restore β-oxidation by enhancing the expression of Trx, GSR, Srx1, Prx3, and GPx. However, Nrf2 also participates in regulating the occurrence of mitophagy and aggravating ROS accumulation by reducing the expression of TXNIP and increasing the combination of Trx and ASK1.The abbreviations in Figure 3 are defined as follows: DNL (de novo lipogenesis); VLDL (very-low-density lipoprotein); MPTP (mitochondrial permeability transition pore); CPT1C (carnitine palmitoyl transferase 1C); CPT2C (carnitine palmitoyl transferase 2C); MCAD (medium-chain acyl-CoA dehydrogenase); LCAD (long-chain acyl-CoA dehydrogenase); VLCAD (very-long-chain acyl-CoA dehydrogenase); AMPT/MTOR (adenosine monophosphate-activated protein kinase/mammalian target of rapamycin); BAT (brown adipose tissue); NK-κB (nuclear factor kappa-light-chain-enhancer of activated B cells); LPS (lipopolysaccharide); IKK (IκB kinase); MnSOD (manganese superoxide dismutase); ETC (electron transport chain); TCA (tricarboxylic acid cycle): NADH/FADH2 (nicotinamide adenine dinucleotide (reduced form)/flavin adenine dinucleotide (reduced form)); Nrf2 (nuclear factor erythroid 2-related factor 2); PGAM5 (phosphoglycerate mutase family member 5); Prx3 (peroxiredoxin 3); TXNIP (thioredoxin-interacting protein); and ASK1 (apoptosis signal-regulating kinase).
1.7 Gut Microbiota in the Liver–Gut Axis Influence Mitochondrial Function
The gut–liver axis is a term used to describe the interactions between the liver and resident gut microbiota in the gastrointestinal tract. Recent evidence has shown that there is a related relationship between intestinal microbiota disturbance and mitochondrial dysfunction during the development of MAFLD [
115,
116]. Patients with MAFLD caused by malnutrition have increased permeability of the intestinal epithelial barrier, changes in the proportion of intestinal microbial species, and bacterial translocation in the intestine, leading to abnormal liver inflammation and oxidative reactions, ultimately aggravating the development of MAFLD [
24,
117]. In the MAFLD mouse model, bacterial translocation, such as that of
Helicobacter pylori, and intestinal flora metabolites (endogenous ethanol, etc.) promote the activation of neutrophils, HSCs, and Kupffer cells to produce ROS and secrete peroxidase, chemokines, proinflammatory factors, and corticosterone, thereby exacerbating hepatic steatosis and inflammation [
118,
119].
The gut microbiota affects the host’s synthesis of antioxidants such as glutathione. Mardinoglu, A. et al. found that the antioxidant glutathione was deficient in patients with T2D, thereby causing the accumulation of mt-ROS to aggravate oxidative stress in the liver [
120]. Neish, A.S. et al. found that commensal Lactobacillus could enhance NOX family enzyme activity to promote the production of nonmitochondrial ROS, ultimately affecting the process of systemic metabolic reactions [
121]. Juarez-Fernandez, M. et al. found that methylation-controlled J protein (MCJ) knockout MASH mice fed a high-fat diet had a milder degree of liver damage than WT mice. Then, through intestinal flora transplantation (FMT) into germ-free (GF) mice, they found that the proportion and formation of microorganisms and short-chain fatty acids (SCFAs) in the gut–liver axis in GF mice were improved, increasing the capacity of fatty acid oxidation in GF mice [
122].
In addition, a damaged intestinal barrier is an important prerequisite for the transfer of potentially harmful bacteria and their effector molecules (PAMPs) to the liver. For example, endotoxin is a kind of LPS and a component of commensal Gram-negative bacteria. MASH patients had higher liver endotoxin levels than patients with simple steatosis [
123]. Studies have found that endotoxin can mediate the NF-κB or JNK pathway to regulate cellular oxidative reactions [
124]. Endotoxin can also aggravate steatosis by activating TLR and NOD-like receptors (NLRs) to produce inflammatory factors and chemokines to inhibit the decomposition of FFAs [
125]. Other studies have found that excessive fructose could increase the risk of causing MAFLD, and MASH mice fed excessive fructose had an increased incidence of endotoxemia [
126,
127]. In fructose-treated CYP2E1-null mice, endotoxemia, inflammatory cell infiltration, and ROS production were less significantly increased than those in wild-type mice [
127]. In addition, HepG2 cells treated with LPS could significantly downregulate the activities of catalase and SOD, aggravating the state of oxidative stress [
128]. Above all, the relationship between oxidative stress and endotoxin regulation in MAFLD is complex. On the one hand, oxidative stress can increase intestinal permeability, leading to more endotoxin transfer into the blood and liver, and finally exacerbating liver inflammation and fibrosis. On the other hand, endotoxin can induce oxidative stress in the liver, further damaging hepatocytes by activating the NLRP3 inflammasome and releasing proinflammatory factors such as IL-1β [
129].
Moreover, gut microbiota metabolites can also regulate mitochondrial structure. For instance, Zhao, T. et al. found that butyrate, an intestinal flora metabolite, can significantly alleviate mt-ROS copies caused by a high-sugar diet in db/db mice, reduce their number and content, and effectively restore mitochondrial membrane potential and ATP synthesis capacity [
130]. Previous studies have shown that Bacteroidetes and Firmicutes promote an increase in the level of SCFAs, which are used by mitochondria to synthesize ATP [
117]. Butyrate can also reverse the decrease in the activity of fatty acid β-oxidation rate-limiting enzymes such as CPT1A and acetyl-CoA carboxylase α (ACACα) to improve fatty acid metabolism and thereby reduce the degree of hepatocellular hypertrophy and steatosis [
130]
This entry is adapted from the peer-reviewed paper 10.3390/ijms242417514