1. Introduction
The recently published guidelines defined cardiomyopathy as “a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease, and congenital heart disease sufficient to cause the observed myocardial abnormalities” [
1]. This definition applies to both children and adults and makes no a priori assumptions about aetiology (which can be familial/genetic or acquired) or myocardial pathology.
Currently, cardiomyopathies are distinguished into hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (ACM), restrictive cardiomyopathy (RCM) and a novel entity that has been recently defined as non-dilated left ventricular cardiomyopathy [
1].
RCM characterized by restrictive cardiac physiology is caused by increased myocardial stiffness and a pathological rise in ventricular filling pressure [
2]. The term restrictive derives from Latin
restrictus, meaning tight or limiting the expansion; it is important to recognize that the term “restrictive cardiomyopathy” does not only represent a diagnostic phenotype but also describes the underlying physiology.
Since RCM is rare, clinicians involved in the diagnostic process and management of paediatric patients with RCM might not be as confident as in other more prevalent cardiomyopathies. Hence, doubts are common. Here, the most frequently asked questions posed by the initial evaluation and management of patients with RCM are addressed in a Q&A format.
2. How Common Is RCM?
Among the paediatric population, RCM is the least common cardiomyopathy and is typically diagnosed between the ages of 6–10 years but can present in early infancy or in adulthood, with an approximately equal sex distribution. Aetiologies vary by age and country of origin, with wide geographical differences [
3]. The Pediatric Cardiomyopathy Registry in North America estimated the incidence of RCM at 0.03–0.04 per 100,000 children, accounting for approximately 4% of paediatric cardiomyopathies [
4]. Meanwhile, the incidence of paediatric RCM may be higher in tropical areas of Africa, Asia and South America, where endemic forms are present [
5]. The reason for an increased prevalence of RCM in the tropics is currently under scrutiny. Despite many hypotheses, the exact causes and mechanisms remain unclear. Genetics is a potential factor due to ethnic clustering, but it is difficult to tease out the confounding shared environmental exposures thought to play a role in the development of RCM in tropical areas [
6]. For example, several infectious organisms have been hypothesised to elicit an exaggerated immune response [
7].
3. Is Family History and Genetic Testing Important?
Family history of cardiomyopathy should always be investigated, as nearly 25% of RCM patients have at least one affected relative, with an even higher proportion (42%) among those with a mixed restrictive and hypertrophic phenotype [
8]. This highlights the importance of a detailed family tree, genetic counselling and cascade genetic testing to identify patients at risk of developing RCM or other overlapping phenotypes.
4. What Are the Main Clinical Signs and Symptoms of Paediatric RCM?
Clinical manifestations range from lack of symptoms to overt heart failure (HF); the most common scenario is poor exercise tolerance and fatigue due to low cardiac output [
9]. Since restrictive left ventricle (LV) cannot adjust volumes during exercise, the only way to increase output is to augment heart rate.
Symptoms of HF are nonspecific, challenging to interpret and vary with the age of onset, usually manifesting as difficulty in feeding, tachypnoea, sinus tachycardia and diaphoresis in the infant. Children with RCM frequently have a history of “recurrent lower respiratory tract infections,” wheezing or persistent cough at night [
5]. Adolescence may present with fatigue, dyspnoea or abdominal pain [
10].
On examination, jugular venous distention, hepatomegaly, a loud pulmonary component of second heart sound, gallop rhythm and a systolic murmur due to atrio-ventricular (AV) valve regurgitation may be present. An abnormally high respiratory rate in the newborn, puffy eyelids and sacral oedema are signs of systemic venous congestion, while ankle oedema, which is commonly seen in adults, is not found in infants [
11,
12].
5. When Should I Suspect Restrictive Phenotype on Electrocardiogram or Imaging Techniques?
In the paediatric setting, diagnosing RCM at an early stage is challenging as specific paediatric guidelines for evaluating heart remodelling are missing [
13]. Electrocardiogram (ECG), chest X-ray, echocardiography and NTproBNP are first line evaluations helpful in the rule-in/rule-out process for RCM diagnosis [
14] (
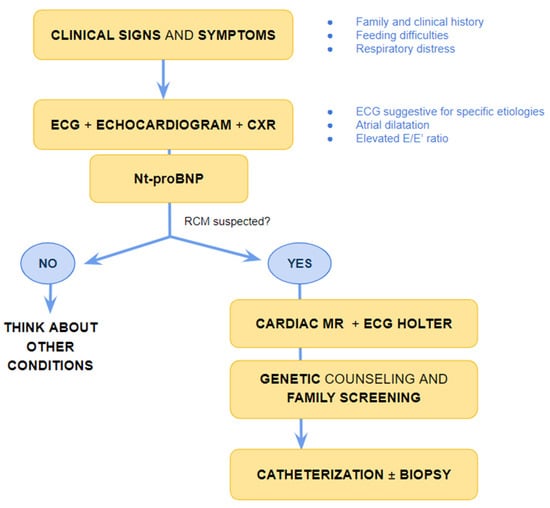
Figure 1).
Figure 1. Suggested flow-chart for initial approach to patient with heart failure and suspected restrictive cardiomyopathy. CXR chest X-ray, ECG electrocardiogram; MR magnetic resonance, Nt-pro-BNP N-terminal pro B type Natriuretic Peptide, RCM-restrictive cardiomyopathy.
Evidence of atrial dilatation on ECG is one of the red flags suggestive of possible RCM, emphasizing the underlying atrial remodelling (
Figure 2) [
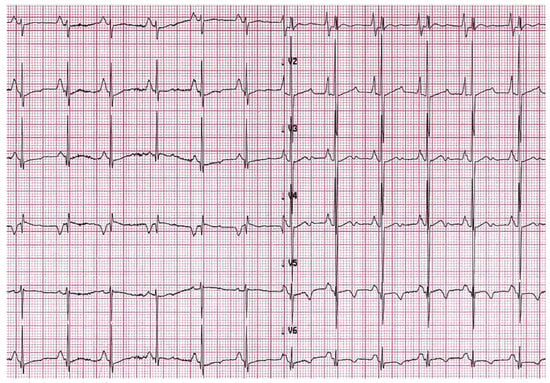
15]. Atrial dilatation reflects increased filling pressures and may predispose to atrial fibrillation in adults with RCM, while this is uncommon in children. Other arrhythmias such as atrio-ventricular (AV) block or intraventricular conduction delay are rare but may develop in the context of specific aetiologies associated with RCM, as discussed below.
Figure 2. A 6-year-old boy with restrictive cardiomyopathy. Electrocardiogram shows abnormal P waves suggestive of atrial enlargement and abnormal ventricular repolarization suggestive of biventricular pressure overload.
Echocardiographic signs reflect the increased pressure overload due to increased stiffness of the ventricles. While systolic function is commonly preserved, initial signs of diastolic dysfunction remain challenging to detect due to the lack of sensitive cutoff values in the paediatric population [
13]. Tissue Doppler imaging is helpful in defining the diastolic function. In a comparison study with invasive hemodynamic measurements, the pulsed Doppler cutoff values of lateral a’ velocity ≤ 4 cm/s and pulmonary vein A wave duration of more than 156 milliseconds showed high sensitivity and specificity in detecting early RCM [
16].
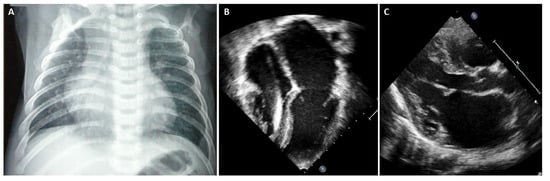
In advanced stages of disease, atrial dilatation is common (Figure 3), pericardial effusion may develop as hemodynamic consequences of increased cardiac filling pressure and Doppler evaluation shows increased early filling velocity (E wave)/atrial filling velocity (A wave) ratio, reduced E wave deceleration time and elevated E/e’ ratio on tissue Doppler.
Figure 3. (A) Chest X-ray, 8-year-old boy with hypertrophic-restrictive cardiomyopathy and congestive heart failure. (B) Echocardiography apical 4-chambers view, 11-year-old girl with restrictive cardiomyopathy, normal ventricular volumes, preserved ejection fraction and severe atrial dilatation. (C) Echocardiography parasternal long axis view, 6-year-old boy with restrictive cardiomyopathy associated to severe left atrial dilatation.
Chest X-ray is often helpful when HF signs are present. In overt RCM phenotype, chest radiography of patients with RCM shows cardiac enlargement due to atrial and pulmonary artery dilatation. In addition, signs of pulmonary congestion and pleural effusion might be present [
17].
Cardiac magnetic resonance (CMR) can be of great importance in the diagnostic workup (
Figure 1). However, the evaluation of diastolic dysfunction at CMR is still based on indirect parameters such as atrial enlargement, T1 mapping and the extension of late gadolinium enhancement (LGE) [
18,
19].
CMR can provide additional information regarding tissue characterization, including the extension of fibrosis, which is an important risk factor for adverse outcomes, an indirect marker of increased wall stiffness [
20].
Besides the general considerations of the imaging findings of RCM, there are specific CMR findings that help in the diagnostic process for specific aetiologies and in the therapeutic management.
Moreover, CMR is helpful in guiding the clinician in differential diagnosis between RCM and constrictive pericarditis, which sometimes shows similar clinical manifestations. However, constrictive pericarditis is typically characterized by pericardial thickening, ventricular interdependence and pericardial LGE when inflammation or extensive fibrosis is present [
21].
6. What Are the Limitations of ECG and Imaging Techniques?
Diagnosis of RCM is often challenging, especially in children. Non-invasive tests such as ECG, echocardiography and CMR can provide valuable information and are often the first steps in the diagnostic work-up of RCM, but they may have limitations that need to be considered.
ECG abnormalities might be subtle or absent in the early phase of the disease. In echocardiography, the reliability of diastolic measurements in children are challenging, given the higher heart rates and limited data on the relationships among the diastolic variables and the degree of dysfunction [
22].
CMR have limitations due to its centre availability, need of sedation and the requirement to be performed in specialized centres [
23].
Overall, none of the non-invasive evaluation can provide a reliable evaluation of the intracardiac pressure, while cardiac catheterization represents the gold standard for the evaluation of intracardiac pressure, the diagnosis of RCM and the reassessment of the patients during follow-up to define the management and therapeutic approach.
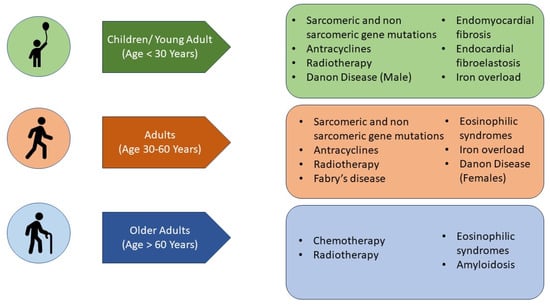
7. What Are the Aetiologies That Mainly Affect the Paediatric Population (Figure 4)?
Endomyocardial fibrosis is considered the most common cause of RCM worldwide, affecting more than 12 million people worldwide [
7], mainly in tropical and subtropical areas. Endomyocardial fibrosis often affects children and young adults belonging to the poorest groups of the population [
24]. It is characterized by recurrent hot phases with inflammation and eosinophilia, progressing to a chronic phase with fibrosis of the ventricular endocardium and sub-endocardium that extends from the apex upwards, often involving the AV valves. A typical CMR pattern is characterized by presence of myocardial oedema during inflammatory phases and/or associated with fibrosis [
25]. The fibrous tissue markedly diminishes the volume and compliance of affected chambers determining a restrictive physiology.
Although the aetiology is unknown, several hypotheses linking infectious agents, toxins or environmental factors to the unique geographical distribution are commonly suggested as possible favouring factors.
Endocardial fibroelastosis is primarily a disease of infants and children, but can rarely present in adults as well [
26]. Due to the rarity of this condition, there are not enough data on the incidence and prevalence. The usual age of presentation is the first year of life [
27], and it is frequently associated with other congenital heart disease as aortic stenosis and hypoplastic left heart syndrome [
28]; nonetheless, the exact aetiology is unknown.
Figure 4. Main aetiologies of restrictive cardiomyopathy according to the age of disease presentation.
The underlying pathophysiology of endocardial fibroelastosis is the deposition of acellular fibrocartilaginous tissue in the subendothelial layer of the endocardium predominantly involving the inflow tracts and apices of ventricles [
29], and intraventricular thrombi formation are an additional complication that CMR can detect [
30].
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics13243666