The NF-κB pathway, first identified in 1986 by Sen and Baltimore, plays a pivotal role in the immune response, cell proliferation, and apoptosis. Its molecular architecture includes five DNA-binding members: REL (c-REL), RELA (p65), RELB, NF-κB1 (p50), and NF-κB2 (p52), with the unique attribute of NF-κB2 (p52) lacking transactivation domains. NF-κB signaling encompasses three distinct pathways: canonical, non-canonical, and atypical, each with unique activation mechanisms and cellular responses. Through multiple graphic depictions, the reference clearly presented the traditional pathways and components of NF-κB. The canonical pathway, generally activated by microbial infections or pro-inflammatory cytokines, involves the phosphorylation and subsequent degradation of IκB proteins by the IκB kinase (IKK) complex, releasing p65/p50 NF-κB dimers for nuclear translocation and transcription activation. The non-canonical pathway, selectively activated by receptors like CD40, B-cell-activating factor receptor (BAFF-R), and lymphotoxin beta receptor (LTβR), primarily involves NF-κB2 (p100/p52) proteins and RELB. This pathway initiates with ligand binding, triggering NF-κB-inducible kinase (NIK) to phosphorylate and activate IKK1 (IKKα), leading to p100’s processing into p52 and the subsequent translocation of p52/RELB dimers to the nucleus, thus regulating gene expression differently compared to the canonical pathway. The atypical pathway, which is less well-characterized, can be triggered by DNA-damaging agents independently of IKK, illustrating the versatility and complexity of NF-κB signaling in cellular dynamics.
1. The Roles of NF-κB in TAMs of GBM
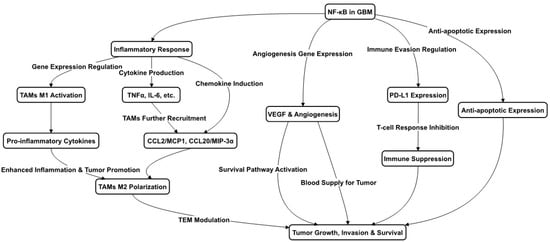
NF-κB is commonly upregulated in various GBM cell types, playing a pivotal role in several critical processes. These include not only traditional immune and inflammatory responses, such as the recruiting and aggregation of microglia and macrophages, but also tumor cell survival and anti-apoptosis. These effects are mediated through the upregulation of NF-κB target genes, which encode growth factors, cytokines, and enzymes [
19], thereby facilitating tumor cell migration and invasion. Moreover, NF-κB has been implicated in the transition of GBM cells from less aggressive phenotypes, such as the proneural phenotype to the more aggressive mesenchymal phenotype. Beyond these roles, NF-κB’s role in GBM extends to promoting angiogenesis and invasiveness. For instance, the overexpression of Bmi-1 in glioma cells has been linked to increased angiogenesis through the upregulation of vascular endothelial growth factor C (VEGF-C) and NF-κB activation [
21]. The experimental use of a non-degradable mutant IκBα, which impedes NF-κB activity, has been observed to curtail the expression of angiogenic molecules and diminish angiogenesis in human glioma xenografts [
22]. These findings underscore NF-κB’s multifaceted role in bolstering tumor aggressiveness and progression, rendering it a promising target for therapeutic intervention. All these functions are depicted in
Figure 1.
Figure 1. NF-κB Pathways in Glioblastoma. This figure outlines the role of NF-κB in GBM progression, highlighting its involvement in inflammation, immune cell dynamics, and tumor survival. NF-κB drives the transition of macrophages to M1 and M2 types, influencing tumor growth and defense mechanisms. It affects cytokine and chemokine levels, aiding in the recruitment and regulation of cells within the tumor environment. Additionally, NF-κB activity contributes to new blood vessel formation and the suppression of the immune response, promoting tumor resilience.
In the context of TAMs within GBM, NF-κB is implicated in resistance to various treatments and the suppression of the immune response. TAM polarization in GBM, encompassing both M1 and M2 states, is significantly influenced by NF-κB. While M1 TAMs promote antitumor responses, M2 TAMs are associated with tumor growth and immune suppression [
23]. Predominantly, GBM TAMs display an M2 phenotype with downregulated NF-κB expression, contributing to the immunosuppressive tumor microenvironment (TME) [
24]. This includes inhibiting T cell activation and functionality, which is essential for an effective antitumor immune response. Moreover, NF-κB is involved in programmed cell death-ligand1 (PD-L1) expression regulation in GBM cells by initiating PD-L1 gene transcription via promoter binding [
25], and indirectly influencing PD-L1 transcriptionally, further contributing to T cell inhibition. The NF-κB signaling modulation of cytokines and chemokines delineates its critical role in reshaping the immune landscape to favor tumor growth.
Strategies aimed at reprogramming TAMs towards an M1 phenotype are thought to be advantageous for patient prognosis. Despite the prevalence of the M2 phenotype during GBM progression, a small fraction of TAMs maintains an M1 status. The presence of necrotic cells within GBM stimulates the expression of TAM chemokines, like the C–C motif chemokine ligand 2/monocyte chemoattractant protein-1 (CL2/MCP-1) and CCL20/MIP-3α (macrophage inflammatory protein 3 alpha), via NF-κB activation, enhancing microglia and macrophage infiltration and attenuating inflammation and apoptosis within the tumor [
26], as shown in
Figure 1. Innovative treatments like proton irradiation have shown promise in reprogramming M2-like TAMs towards an M1 antitumor phenotype [
27], and targeting markers associated with M2-TAMs, such as Arg1 and PD-1, has the potential for reactivating T cell responses and reshaping the TME [
28]. Focusing on the NF-κB p50 transcription factor presents another therapeutic avenue. Experiments in which GL261-Luc glioblastoma cells were implanted in both wild-type and p50 knockout (ko) mice resulted in extended survival in the ko mice, characterized by a shift in TAM populations towards pro-inflammatory M1 markers and a threefold increase in tumor-associated CD4 T cells compared to wild-type mice [
29]. Ongoing research is critical to elucidate NF-κB’s intricate roles across the spectrum of M1 and M2 macrophage and microglia phenotypes in glioblastoma.
2. The Roles of NF-κB in Microglia/Macrophges of AD
In AD, the NF-κB pathway is a pivotal mediator in the functional dynamics of microglia and macrophages. The canonical NF-κB pathway, predominantly involving the p65 (RelA) and p50 subunits, is typically associated with the classical M1 activation [
30]. This pathway is engaged by pro-inflammatory stimuli, such as lipopolysaccharide (LPS) and cytokines including TNFα and IL-1β, as well as iNOS—a marker commonly attributed to the M1 microglial phenotype. Upon activation, NF-κB orchestrates the microglial release of pro-inflammatory mediators, including IL-1, IL-6, tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), and CCL2. These cytokines contribute to neuronal and oligodendrocyte injury, thereby accelerating AD pathogenesis [
31]. Consistently, mice lacking the NF-κB1 (p50) subunit (lacking the repressor protein) exhibited enhanced NF-κB activity, leading to more microglia, chronic inflammation, and early onset memory loss, whereas the anti-inflammatory treatment reduced inflammation and restored memory loss [
32]. An early event in AD development is microglial activation, where NF-κB signaling in reactive microglia is a key contributor to the sustained inflammation characteristic of the disease. Contrary to the M1 profile, microglia in a traditional ‘M2’ state respond to signals of tissue injury by assuming an amoeboid morphology and migrating to injury sites to phagocytose cellular debris and combat pathogens, thus preserving CNS integrity [
33]. Although historically depicted as a binary M1/M2 system—a concept borrowed from macrophage biology—the classification of microglial phenotypes has since evolved. This binary framework is now regarded as overly reductive, particularly within the context of neurodegenerative conditions like AD, and is addressed in depth in a subsequent section (
Here should point out that the subsequence section in the original paper.).Nevertheless, therapeutic targeting of the NF-κB pathway in M1 microglia presents a viable strategy for modulating the microglial phenotype balance, offering potential for AD treatment [
34,
35]. For instance, studies of a microglial BV2 cell culture demonstrated that neuregulin-1 modulates microglial activation and phenotype shifting by inhibiting the NF-κB pathway, likely via an ErbB4-dependent mechanism [
35,
36]. Similarly, research on Sarcodonin A—a natural compound extracted from mushrooms—highlight its neuroprotective potential by attenuating M1 polarization in microglia, reinforcing its candidacy for AD therapeutics targeting microglial neuroinflammation [
35]. Recent findings also underscore the role of Dectin-1, a pattern recognition receptor on microglia, in mediating the inflammatory response to amyloid-beta (Aβ) in AD [
37]. The direct interaction between Aβ and Dectin-1 triggers signaling through NF-κB, while Dectin-1 knockout models exhibit a reduction in Aβ-induced microglial activation and inflammation. Moreover, AD brains have shown an upregulation of NF-κB-regulated miRNAs, establishing a gene expression program that correlates with AD pathology, which will be elaborated upon in the following sections. The critical involvement of NF-κB in microglia is further corroborated by the localization of numerous AD risk loci within or adjacent to genes predominantly expressed in microglia, signifying their foundational role in early disease stages. Collectively, these findings bolster the hypothesis that NF-κB targeting in M1 microglia may yield therapeutic advantages in AD.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010016