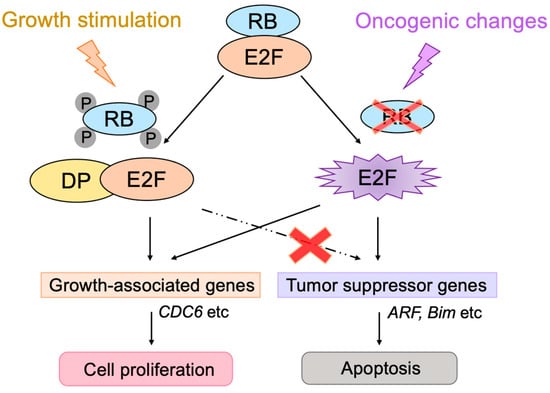
The transcription factor E2F links the RB pathway to the p53 pathway upon loss of function of pRB, thereby playing a pivotal role in the suppression of tumorigenesis. E2F fulfills a major role in cell proliferation by controlling a variety of growth-associated genes. The activity of E2F is controlled by the tumor suppressor pRB, which binds to E2F and actively suppresses target gene expression, thereby restraining cell proliferation. Signaling pathways originating from growth stimulative and growth suppressive signals converge on pRB (the RB pathway) to regulate E2F activity. In most cancers, the function of pRB is compromised by oncogenic mutations, and E2F activity is enhanced, thereby facilitating cell proliferation to promote tumorigenesis. Upon such events, E2F activates the Arf tumor suppressor gene, leading to activation of the tumor suppressor p53 to protect cells from tumorigenesis. ARF inactivates MDM2, which facilitates degradation of p53 through proteasome by ubiquitination (the p53 pathway). P53 suppresses tumorigenesis by inducing cellular senescence or apoptosis. Hence, in almost all cancers, the p53 pathway is also disabled. In this review, we will summarize the canonical RB-E2F-p53 pathway and introduce non-classical functions of each component, which may be relevant to cancer biology.
- RB
- E2F
- ARF
- MDM2
- p53
1. Non-Classical Functions of Each Component of the RB-E2F-ARF-MDM2-p53 Pathway
2. Non-Classical Functions of RB
3. Non-Classical Functions of E2F
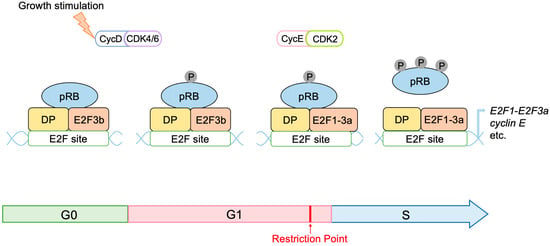
3.1. Unique Properties of E2F in Linking the RB Pathway to the p53 Pathway
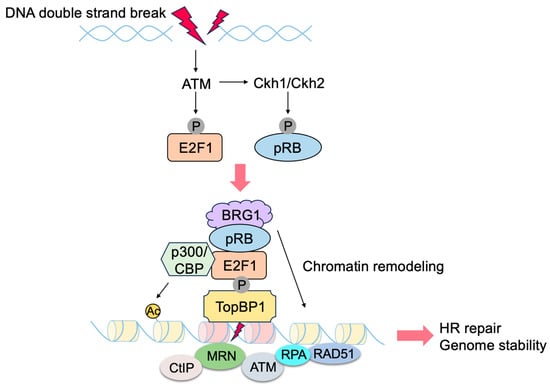
3.2. E2F1 Targets Involved in p53-Independent Pathways for the Induction of Apoptosis
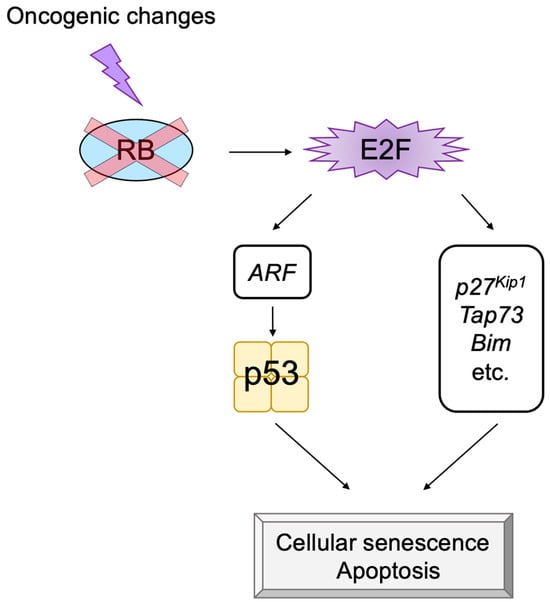
3.3. Cancer-Cell-Specific Deregulated E2F Activity as a Cancer-Cell-Specific Targeting Tool
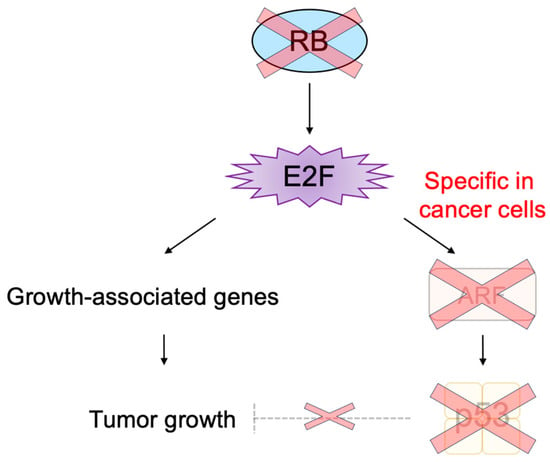
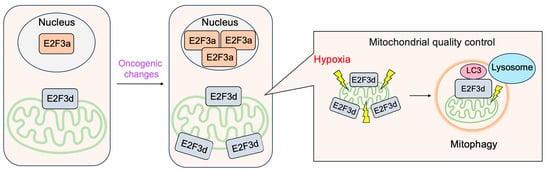
3.4. E2F3d, a Novel Member of the E2F3 Family, Mediates Hypoxia-Induced Mitophagy in Cancer Cells
4. p53-Independent Functions of ARF in Tumor Suppression
4.1. ARF Suppresses Ribosomal Biogenesis
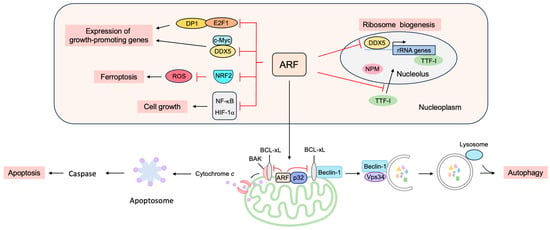
4.2. ARF Suppresses the Expression of Growth-Promoting Genes
4.3. ARF Facilitates Apoptosis at the Mitochondria
4.4. ARF Facilitates Autophagy at the Mitochondria
4.5. ARF Contributes to Genome Stability
4.6. ARF Facilitates the SUMOylation of Interacting Proteins
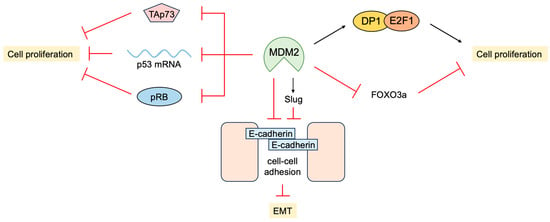
5. P53-Independent Functions of MDM2 in Tumor Promotion
6. Non-Classical Functions of p53
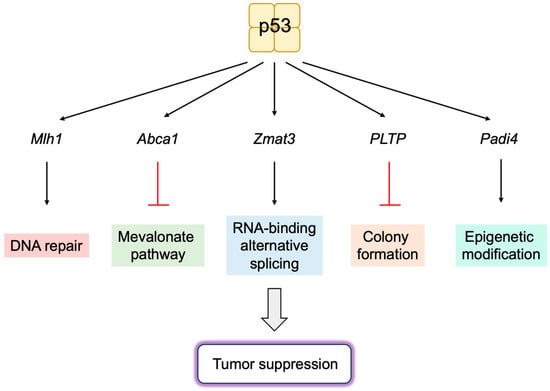
6.1. Novel p53 Targets Genes Important for Tumor Suppression
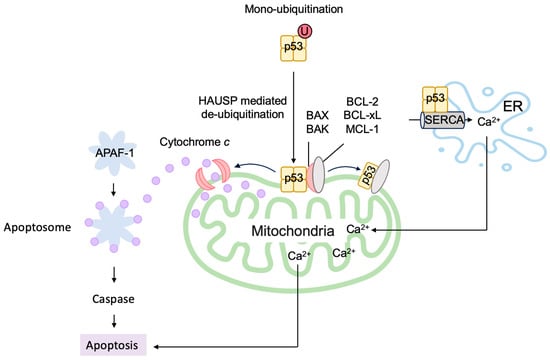
6.2. P53 Directly Induces Apoptosis at the Mitochondria
6.3. P53 Induces Autophagy to Suppress Tumorigenesis
This entry is adapted from the peer-reviewed paper 10.3390/biology12121511
References
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872.
- Biswas, A.K.; Johnson, D.G. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012, 72, 13–17.
- Velez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776.
- Manickavinayaham, S.; Dennehey, B.K.; Johnson, D.G. Direct Regulation of DNA Repair by E2F and RB in Mammals and Plants: Core Function or Convergent Evolution? Cancers 2021, 13, 934.
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844.
- Liu, K.; Lin, F.T.; Ruppert, J.M.; Lin, W.C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 2003, 23, 3287–3304.
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093.
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512.
- Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176.
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514.
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294.
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Bedford, E.; Klein, B.J.; Kutateladze, T.G.; Liu, B.; Bedford, M.T.; Johnson, D.G. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat. Commun. 2019, 10, 4951.
- Choi, E.H.; Kim, K.P. E2F1 facilitates DNA break repair by localizing to break sites and enhancing the expression of homologous recombination factors. Exp. Mol. Med. 2019, 51, 1–12.
- Guo, R.; Chen, J.; Zhu, F.; Biswas, A.K.; Berton, T.R.; Mitchell, D.L.; Johnson, D.G. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J. Biol. Chem. 2010, 285, 19308–19315.
- Biswas, A.K.; Mitchell, D.L.; Johnson, D.G. E2F1 responds to ultraviolet radiation by directly stimulating DNA repair and suppressing carcinogenesis. Cancer Res. 2014, 74, 3369–3377.
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 2013, 25, 735–740.
- Schaal, C.; Pillai, S.; Chellappan, S.P. The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 2014, 121, 147–182.
- Stevaux, O.; Dyson, N.J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 2002, 14, 684–691.
- Manning, A.L.; Dyson, N.J. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011, 21, 433–441.
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226.
- Talluri, S.; Dick, F.A. Regulation of transcription and chromatin structure by pRB: Here, there and everywhere. Cell Cycle 2012, 11, 3189–3198.
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell. Biol. 2013, 14, 297–306.
- Popov, B.; Petrov, N. pRb-E2F signaling in life of mesenchymal stem cells: Cell cycle, cell fate, and cell differentiation. Genes Dis. 2014, 1, 174–187.
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890.
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502.
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451.
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324.
- Komori, H.; Enomoto, M.; Nakamura, M.; Iwanaga, R.; Ohtani, K. Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J. 2005, 24, 3724–3736.
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20.
- Komori, H.; Goto, Y.; Kurayoshi, K.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Araki, K.; Ohtani, K. Differential requirement for dimerization partner DP between E2F-dependent activation of tumor suppressor and growth-related genes. Sci. Rep. 2018, 8, 8438.
- Helin, K.; Wu, C.L.; Fattaey, A.R.; Lees, J.A.; Dynlacht, B.D.; Ngwu, C.; Harlow, E. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 1993, 7, 1850–1861.
- Ozono, E.; Komori, H.; Iwanaga, R.; Ikeda, M.A.; Iseki, S.; Ohtani, K. E2F-like elements in p27(Kip1) promoter specifically sense deregulated E2F activity. Genes Cells 2009, 14, 89–99.
- Ozono, E.; Komori, H.; Iwanaga, R.; Tanaka, T.; Sakae, T.; Kitamura, H.; Yamaoka, S.; Ohtani, K. Tumor suppressor TAp73 gene specifically responds to deregulated E2F activity in human normal fibroblasts. Genes Cells 2012, 17, 660–672.
- Kitamura, H.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Okuno, J.; Shimizu, E.; Kurayoshi, K.; Kugawa, K.; Toh, H.; Ohtani, K. Identification of novel target genes specifically activated by deregulated E2F in human normal fibroblasts. Genes Cells 2015, 20, 739–757.
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000, 407, 645–648.
- Stiewe, T.; Putzer, B.M. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat. Genet. 2000, 26, 464–469.
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112.
- Knudsen, E.S.; Wang, J.Y. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099.
- Laine, A.; Westermarck, J. Molecular pathways: Harnessing E2F1 regulation for prosenescence therapy in p53-defective cancer cells. Clin. Cancer Res. 2014, 20, 3644–3650.
- Tsukuda, K.; Wiewrodt, R.; Molnar-Kimber, K.; Jovanovic, V.P.; Amin, K.M. An E2F-responsive replication-selective adenovirus targeted to the defective cell cycle in cancer cells: Potent antitumoral efficacy but no toxicity to normal cell. Cancer Res. 2002, 62, 3438–3447.
- Dubensky, T.W., Jr. (Re-)Engineering tumor cell-selective replicating adenoviruses: A step in the right direction toward systemic therapy for metastatic disease. Cancer Cell 2002, 1, 307–309.
- Jakubczak, J.L.; Ryan, P.; Gorziglia, M.; Clarke, L.; Hawkins, L.K.; Hay, C.; Huang, Y.; Kaloss, M.; Marinov, A.; Phipps, S.; et al. An oncolytic adenovirus selective for retinoblastoma tumor suppressor protein pathway-defective tumors: Dependence on E1A, the E2F-1 promoter, and viral replication for selectivity and efficacy. Cancer Res. 2003, 63, 1490–1499.
- Johnson, D.G.; Ohtani, K.; Nevins, J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994, 8, 1514–1525.
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. p14ARF links the tumour suppressors RB and p53. Nature 1998, 395, 124–125.
- Kurayoshi, K.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Komori, H.; Ohtani, K. Cancer cell specific cytotoxic gene expression mediated by ARF tumor suppressor promoter constructs. Biochem. Biophys. Res. Commun. 2014, 450, 240–246.
- Nakajima, R.; Zhao, L.; Zhou, Y.; Shirasawa, M.; Uchida, A.; Murakawa, H.; Fikriyanti, M.; Iwanaga, R.; Bradford, A.P.; Araki, K.; et al. Deregulated E2F Activity as a Cancer-Cell Specific Therapeutic Tool. Genes 2023, 14, 393.
- Araki, K.; Kawauchi, K.; Sugimoto, W.; Tsuda, D.; Oda, H.; Yoshida, R.; Ohtani, K. Mitochondrial protein E2F3d, a distinctive E2F3 product, mediates hypoxia-induced mitophagy in cancer cells. Commun. Biol. 2019, 2, 3.
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797.
- Benevolenskaya, E.V.; Frolov, M.V. Emerging links between E2F control and mitochondrial function. Cancer Res. 2015, 75, 619–623.
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338.
- Julian, L.M.; Blais, A. Transcriptional control of stem cell fate by E2Fs and pocket proteins. Front. Genet. 2015, 6, 161.
- Jusino, S.; Saavedra, H.I. Role of E2Fs and mitotic regulators controlled by E2Fs in the epithelial to mesenchymal transition. Exp. Biol. Med. 2019, 244, 1419–1429.
- Xie, D.; Pei, Q.; Li, J.; Wan, X.; Ye, T. Emerging Role of E2F Family in Cancer Stem Cells. Front. Oncol. 2021, 11, 723137.
- Hsu, J.; Sage, J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle 2016, 15, 3183–3190.
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87.
- Kung, C.P.; Weber, J.D. It’s Getting Complicated—A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744.
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000, 14, 2358–2365.
- Eymin, B.; Leduc, C.; Coll, J.L.; Brambilla, E.; Gazzeri, S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835.
- Sandoval, R.; Xue, J.; Pilkinton, M.; Salvi, D.; Kiyokawa, H.; Colamonici, O.R. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J. Biol. Chem. 2004, 279, 32275–32280.
- Muniz, V.P.; Barnes, J.M.; Paliwal, S.; Zhang, X.; Tang, X.; Chen, S.; Zamba, K.D.; Cullen, J.J.; Meyerholz, D.K.; Meyers, S.; et al. The ARF tumor suppressor inhibits tumor cell colonization independent of p53 in a novel mouse model of pancreatic ductal adenocarcinoma metastasis. Mol. Cancer Res. 2011, 9, 867–877.
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424.
- Ayrault, O.; Andrique, L.; Larsen, C.J.; Seite, P. Human Arf tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene 2004, 23, 8097–8104.
- Lessard, F.; Morin, F.; Ivanchuk, S.; Langlois, F.; Stefanovsky, V.; Rutka, J.; Moss, T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell 2010, 38, 539–550.
- Brady, S.N.; Yu, Y.; Maggi, L.B., Jr.; Weber, J.D. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol. Cell. Biol. 2004, 24, 9327–9338.
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol. Cell. Biol. 2004, 24, 985–996.
- Maggi, L.B., Jr.; Kuchenruether, M.; Dadey, D.Y.; Schwope, R.M.; Grisendi, S.; Townsend, R.R.; Pandolfi, P.P.; Weber, J.D. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol. Cell. Biol. 2008, 28, 7050–7065.
- Moulin, S.; Llanos, S.; Kim, S.H.; Peters, G. Binding to nucleophosmin determines the localization of human and chicken ARF but not its impact on p53. Oncogene 2008, 27, 2382–2389.
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271.
- Apicelli, A.J.; Maggi, L.B., Jr.; Hirbe, A.C.; Miceli, A.P.; Olanich, M.E.; Schulte-Winkeler, C.L.; Saporita, A.J.; Kuchenreuther, M.; Sanchez, J.; Weilbaecher, K.; et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol. Cell. Biol. 2008, 28, 1068–1080.
- Saporita, A.J.; Chang, H.C.; Winkeler, C.L.; Apicelli, A.J.; Kladney, R.D.; Wang, J.; Townsend, R.R.; Michel, L.S.; Weber, J.D. RNA helicase DDX5 is a p53-independent target of ARF that participates in ribosome biogenesis. Cancer Res. 2011, 71, 6708–6717.
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; Della Fazia, M.A.; Servillo, G. Hepatocyte odd protein shuttling (HOPS) is a bridging protein in the nucleophosmin-p19 Arf network. Oncogene 2013, 32, 3350–3358.
- Kuchenreuther, M.J.; Weber, J.D. The ARF tumor-suppressor controls Drosha translation to prevent Ras-driven transformation. Oncogene 2014, 33, 300–307.
- Maggi, L.B., Jr.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839.
- Dominguez-Brauer, C.; Brauer, P.M.; Chen, Y.J.; Pimkina, J.; Raychaudhuri, P. Tumor suppression by ARF: Gatekeeper and caretaker. Cell Cycle 2010, 9, 86–89.
- Eymin, B.; Karayan, L.; Seite, P.; Brambilla, C.; Brambilla, E.; Larsen, C.J.; Gazzeri, S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene 2001, 20, 1033–1041.
- Mason, S.L.; Loughran, O.; La Thangue, N.B. p14(ARF) regulates E2F activity. Oncogene 2002, 21, 4220–4230.
- Zhang, H.J.; Li, W.J.; Gu, Y.Y.; Li, S.Y.; An, G.S.; Ni, J.H.; Jia, H.T. p14ARF interacts with E2F factors to form p14ARF-E2F/partner-DNA complexes repressing E2F-dependent transcription. J. Cell Biochem. 2010, 109, 693–701.
- Datta, A.; Nag, A.; Raychaudhuri, P. Differential regulation of E2F1, DP1, and the E2F1/DP1 complex by ARF. Mol. Cell. Biol. 2002, 22, 8398–8408.
- Datta, A.; Sen, J.; Hagen, J.; Korgaonkar, C.K.; Caffrey, M.; Quelle, D.E.; Hughes, D.E.; Ackerson, T.J.; Costa, R.H.; Raychaudhuri, P. ARF directly binds DP1: Interaction with DP1 coincides with the G1 arrest function of ARF. Mol. Cell. Biol. 2005, 25, 8024–8036.
- Martelli, F.; Hamilton, T.; Silver, D.P.; Sharpless, N.E.; Bardeesy, N.; Rokas, M.; DePinho, R.A.; Livingston, D.M.; Grossman, S.R. p19ARF targets certain E2F species for degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 4455–4460.
- Datta, A.; Nag, A.; Pan, W.; Hay, N.; Gartel, A.L.; Colamonici, O.; Mori, Y.; Raychaudhuri, P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J. Biol. Chem. 2004, 279, 36698–36707.
- Tago, K.; Funakoshi-Tago, M.; Itoh, H.; Furukawa, Y.; Kikuchi, J.; Kato, T.; Suzuki, K.; Yanagisawa, K. Arf tumor suppressor disrupts the oncogenic positive feedback loop including c-Myc and DDX5. Oncogene 2015, 34, 314–322.
- Fatyol, K.; Szalay, A.A. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (HIF-1alpha) and inhibits HIF-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429.
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25.
- Rocha, S.; Garrett, M.D.; Campbell, K.J.; Schumm, K.; Perkins, N.D. Regulation of NF-kappaB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005, 24, 1157–1169.
- Hyder, U.; McCann, J.L.; Wang, J.; Fung, V.; Bayo, J.; D’Orso, I. The ARF tumor suppressor targets PPM1G/PP2Cgamma to counteract NF-kappaB transcription tuning cell survival and the inflammatory response. Proc. Natl. Acad. Sci. USA 2020, 117, 32594–32605.
- Kawagishi, H.; Nakamura, H.; Maruyama, M.; Mizutani, S.; Sugimoto, K.; Takagi, M.; Sugimoto, M. ARF suppresses tumor angiogenesis through translational control of VEGFA mRNA. Cancer Res. 2010, 70, 4749–4758.
- Paliwal, S.; Pande, S.; Kovi, R.C.; Sharpless, N.E.; Bardeesy, N.; Grossman, S.R. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol. Cell. Biol. 2006, 26, 2360–2372.
- Kovi, R.C.; Paliwal, S.; Pande, S.; Grossman, S.R. An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ. 2010, 17, 513–521.
- Lu, W.; Xie, Y.; Ma, Y.; Matusik, R.J.; Chen, Z. ARF represses androgen receptor transactivation in prostate cancer. Mol. Endocrinol. 2013, 27, 635–648.
- Herkert, B.; Dwertmann, A.; Herold, S.; Abed, M.; Naud, J.F.; Finkernagel, F.; Harms, G.S.; Orian, A.; Wanzel, M.; Eilers, M. The Arf tumor suppressor protein inhibits Miz1 to suppress cell adhesion and induce apoptosis. J. Cell Biol. 2010, 188, 905–918.
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109.
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e224.
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553.
- Irvine, M.; Philipsz, S.; Frausto, M.; Mijatov, B.; Gallagher, S.J.; Fung, C.; Becker, T.M.; Kefford, R.F.; Rizos, H. Amino terminal hydrophobic import signals target the p14(ARF) tumor suppressor to the mitochondria. Cell Cycle 2010, 9, 829–839.
- Muer, A.; Overkamp, T.; Gillissen, B.; Richter, A.; Pretzsch, T.; Milojkovic, A.; Dorken, B.; Daniel, P.T.; Hemmati, P. p14(ARF)-induced apoptosis in p53 protein-deficient cells is mediated by BH3-only protein-independent derepression of Bak protein through down-regulation of Mcl-1 and Bcl-xL proteins. J. Biol. Chem. 2012, 287, 17343–17352.
- Repenning, A.; Happel, D.; Bouchard, C.; Meixner, M.; Verel-Yilmaz, Y.; Raifer, H.; Holembowski, L.; Krause, E.; Kremmer, E.; Feederle, R.; et al. PRMT1 promotes the tumor suppressor function of p14(ARF) and is indicative for pancreatic cancer prognosis. EMBO J. 2021, 40, e106777.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- Abida, W.M.; Gu, W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357.
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810.
- Pimkina, J.; Murphy, M.E. Interaction of the ARF tumor suppressor with cytosolic HSP70 contributes to its autophagy function. Cancer Biol. Ther. 2011, 12, 503–509.
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565.
- Fontana, R.; Guidone, D.; Angrisano, T.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. Mutation of the Conserved Threonine 8 within the Human ARF Tumour Suppressor Protein Regulates Autophagy. Biomolecules 2022, 12, 126.
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475.
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683.
- Tompkins, V.S.; Hagen, J.; Frazier, A.A.; Lushnikova, T.; Fitzgerald, M.P.; di Tommaso, A.; Ladeveze, V.; Domann, F.E.; Eischen, C.M.; Quelle, D.E. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J. Biol. Chem. 2007, 282, 1322–1333.
- Britigan, E.M.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol. Biol. Cell 2014, 25, 2761–2773.
- Xirodimas, D.P.; Chisholm, J.; Desterro, J.M.; Lane, D.P.; Hay, R.T. P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Lett. 2002, 528, 207–211.
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694.
- Calabro, V.; Mansueto, G.; Santoro, R.; Gentilella, A.; Pollice, A.; Ghioni, P.; Guerrini, L.; La Mantia, G. Inhibition of p63 transcriptional activity by p14ARF: Functional and physical link between human ARF tumor suppressor and a member of the p53 family. Mol. Cell. Biol. 2004, 24, 8529–8540.
- Vivo, M.; Di Costanzo, A.; Fortugno, P.; Pollice, A.; Calabro, V.; La Mantia, G. Downregulation of DeltaNp63alpha in keratinocytes by p14ARF-mediated SUMO-conjugation and degradation. Cell Cycle 2009, 8, 3545–3551.
- Ranieri, M.; Vivo, M.; De Simone, M.; Guerrini, L.; Pollice, A.; La Mantia, G.; Calabro, V. Sumoylation and ubiquitylation crosstalk in the control of DeltaNp63alpha protein stability. Gene 2018, 645, 34–40.
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279.
- Kuo, M.L.; den Besten, W.; Thomas, M.C.; Sherr, C.J. Arf-induced turnover of the nucleolar nucleophosmin-associated SUMO-2/3 protease Senp3. Cell Cycle 2008, 7, 3378–3387.
- Xiao, Z.X.; Chen, J.; Levine, A.J.; Modjtahedi, N.; Xing, J.; Sellers, W.R.; Livingston, D.M. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 1995, 375, 694–698.
- Hsieh, J.K.; Chan, F.S.; O’Connor, D.J.; Mittnacht, S.; Zhong, S.; Lu, X. RB regulates the stability and the apoptotic function of p53 via MDM2. Mol. Cell 1999, 3, 181–193.
- Martin, K.; Trouche, D.; Hagemeier, C.; Sorensen, T.S.; La Thangue, N.B.; Kouzarides, T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature 1995, 375, 691–694.
- Loughran, O.; La Thangue, N.B. Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol. Cell. Biol. 2000, 20, 2186–2197.
- Lundgren, K.; Montes de Oca Luna, R.; McNeill, Y.B.; Emerick, E.P.; Spencer, B.; Barfield, C.R.; Lozano, G.; Rosenberg, M.P.; Finlay, C.A. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997, 11, 714–725.
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105.
- Gajjar, M.; Candeias, M.M.; Malbert-Colas, L.; Mazars, A.; Fujita, J.; Olivares-Illana, V.; Fahraeus, R. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 2012, 21, 25–35.
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63.
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189.
- Bouska, A.; Eischen, C.M. Mdm2 affects genome stability independent of p53. Cancer Res. 2009, 69, 1697–1701.
- Yang, J.Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.C. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol. 2006, 26, 7269–7282.
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148.
- Jung, C.H.; Kim, J.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Mdm2 increases cellular invasiveness by binding to and stabilizing the Slug mRNA. Cancer Lett. 2013, 335, 270–277.
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704.
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G., Jr.; Oren, M.; Chen, J.; Lu, H. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999, 19, 3257–3266.
- Balint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929.
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y. MDM2 and MDMX bind and stabilize the p53-related protein p73. Curr. Biol. 1999, 9, 829–832.
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591.
- Klein, A.M.; Biderman, L.; Tong, D.; Alaghebandan, B.; Plumber, S.A.; Mueller, H.S.; van Vlimmeren, A.; Katz, C.; Prives, C. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc. Natl. Acad. Sci. USA 2021, 118, e2102420118.
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023, 13, 1210–1229.
- Valente, L.J.; Gray, D.H.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345.
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113.
- Janic, A.; Valente, L.J.; Wakefield, M.J.; Di Stefano, L.; Milla, L.; Wilcox, S.; Yang, H.; Tai, L.; Vandenberg, C.J.; Kueh, A.J.; et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat. Med. 2018, 24, 947–953.
- Mello, S.S.; Valente, L.J.; Raj, N.; Seoane, J.A.; Flowers, B.M.; McClendon, J.; Bieging-Rolett, K.T.; Lee, J.; Ivanochko, D.; Kozak, M.M.; et al. A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 2017, 32, 460–473.e466.
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e519.
- Jiang, D.; Brady, C.A.; Johnson, T.M.; Lee, E.Y.; Park, E.J.; Scott, M.P.; Attardi, L.D. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. USA 2011, 108, 17123–17128.
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583.
- Bieging-Rolett, K.T.; Kaiser, A.M.; Morgens, D.W.; Boutelle, A.M.; Seoane, J.A.; Van Nostrand, E.L.; Zhu, C.; Houlihan, S.L.; Mello, S.S.; Yee, B.A.; et al. Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol. Cell 2020, 80, 452–469.e459.
- Muys, B.R.; Anastasakis, D.G.; Claypool, D.; Pongor, L.; Li, X.L.; Grammatikakis, I.; Liu, M.; Wang, X.; Prasanth, K.V.; Aladjem, M.I.; et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev. 2021, 35, 102–116.
- Gnanapradeepan, K.; Indeglia, A.; Stieg, D.C.; Clarke, N.; Shao, C.; Dougherty, J.F.; Murali, N.; Murphy, M.E. PLTP is a p53 target gene with roles in cancer growth suppression and ferroptosis. J. Biol. Chem. 2022, 298, 102637.
- Indeglia, A.; Leung, J.C.; Miller, S.A.; Leu, J.I.; Dougherty, J.F.; Clarke, N.L.; Kirven, N.A.; Shao, C.; Ke, L.; Lovell, S.; et al. An African-Specific Variant of TP53 Reveals PADI4 as a Regulator of p53-Mediated Tumor Suppression. Cancer Discov. 2023, 13, 1696–1719.
- Wang, C.; Yang, Y.; Wu, X.; Li, J.; Liu, K.; Fang, D.; Li, B.; Shan, G.; Mei, X.; Wang, F.; et al. Reciprocal modulation of long noncoding RNA EMS and p53 regulates tumorigenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2111409119.
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283.
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell Oncol. 2014, 1, e955995.
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 2000, 275, 16202–16212.
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590.
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014.
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606.
- Hagn, F.; Klein, C.; Demmer, O.; Marchenko, N.; Vaseva, A.; Moll, U.M.; Kessler, H. BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J. Biol. Chem. 2010, 285, 3439–3450.
- Wei, H.; Qu, L.; Dai, S.; Li, Y.; Wang, H.; Feng, Y.; Chen, X.; Jiang, L.; Guo, M.; Li, J.; et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 2021, 12, 2280.
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450.
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934.
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 retains p53 in the cytoplasm and sustains p53-dependent mitochondrial apoptosis. EMBO Rep. 2020, 21, e48073.
- Ferracchiato, S.; Di-Iacovo, N.; Scopetti, D.; Piobbico, D.; Castelli, M.; Pieroni, S.; Gargaro, M.; Manni, G.; Brancorsini, S.; Della-Fazia, M.A.; et al. Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis. Int. J. Mol. Sci. 2021, 22, 7186.
- Sorrentino, G.; Mioni, M.; Giorgi, C.; Ruggeri, N.; Pinton, P.; Moll, U.; Mantovani, F.; Del Sal, G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ. 2013, 20, 198–208.
- Follis, A.V.; Llambi, F.; Merritt, P.; Chipuk, J.E.; Green, D.R.; Kriwacki, R.W. Pin1-Induced Proline Isomerization in Cytosolic p53 Mediates BAX Activation and Apoptosis. Mol. Cell 2015, 59, 677–684.
- Li, L.; Su, Z.; Zou, Z.; Tan, H.; Cai, D.; Su, L.; Gu, Z. Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019, 10, 96.
- Sykes, S.M.; Stanek, T.J.; Frank, A.; Murphy, M.E.; McMahon, S.B. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J. Biol. Chem. 2009, 284, 20197–20205.
- You, H.; Yamamoto, K.; Mak, T.W. Regulation of transactivation-independent proapoptotic activity of p53 by FOXO3a. Proc. Natl. Acad. Sci. USA 2006, 103, 9051–9056.
- Follis, A.V.; Chipuk, J.E.; Fisher, J.C.; Yun, M.K.; Grace, C.R.; Nourse, A.; Baran, K.; Ou, L.; Min, L.; White, S.W.; et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013, 9, 163–168.
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784.
- Sung, Y.J.; Kao, T.Y.; Kuo, C.L.; Fan, C.C.; Cheng, A.N.; Fang, W.C.; Chou, H.Y.; Lo, Y.K.; Chen, C.H.; Jiang, S.S.; et al. Mitochondrial Lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. 2018, 9, 697.
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134.
- Mah, L.Y.; O’Prey, J.; Baudot, A.D.; Hoekstra, A.; Ryan, K.M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 2012, 8, 18–28.
- Mrschtik, M.; Ryan, K.M. Another DRAM involved in autophagy and cell death. Autophagy 2016, 12, 603–605.
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031.
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012, 19, 1525–1535.
- Wu, G.S.; Saftig, P.; Peters, C.; El-Deiry, W.S. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 1998, 16, 2177–2183.
- Ikeguchi, M.; Sakatani, T.; Ueta, T.; Fukuda, K.; Oka, S.; Hisamitsu, K.; Yamaguchi, K.; Tsujitani, S.; Kaibara, N. Correlation between cathepsin D expression and p53 protein nuclear accumulation in oesophageal squamous cell carcinoma. J. Clin. Pathol. 2002, 55, 121–126.
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660.
- Yeo, S.Y.; Itahana, Y.; Guo, A.K.; Han, R.; Iwamoto, K.; Nguyen, H.T.; Bao, Y.; Kleiber, K.; Wu, Y.J.; Bay, B.H.; et al. Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. eLife 2016, 5, e07101.
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350.
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77.
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019, 38, 3729–3742.
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235.
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140.
- Lee, E.F.; Smith, N.A.; Soares da Costa, T.P.; Meftahi, N.; Yao, S.; Harris, T.J.; Tran, S.; Pettikiriarachchi, A.; Perugini, M.A.; Keizer, D.W.; et al. Structural insights into BCL2 pro-survival protein interactions with the key autophagy regulator BECN1 following phosphorylation by STK4/MST1. Autophagy 2019, 15, 785–795.