Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Osteopontin (OPN), an inflammatory cytokine and biomarker of AD, is implicated in Aβ clearance and toxicity, microglial activation, and inflammation, and is considered to be a potential therapeutic target.
- Alzheimer’s disease
- osteopontin
- microglia
1. Introduction
Alzheimer’s disease (AD), the fifth leading global cause of death, is a neurodegenerative condition that, along with subcortical vascular cognitive impairment (SVCI), is responsible for 75% of all dementia cases [1][2][3]. Current estimates place the global population of dementia at 57.4 million, and this is predicted to grow to 152.8 million by the year 2050 in parallel with aging populations, becoming a correspondingly greater public health concern [4][5]. There is no cure for AD, and its progressive course, along with the limited efficacy of current disease-modifying treatments, severely impacts the quality of life of those who are afflicted, with potentially devastating familial, social, and economic consequences. The clinical progression of AD is marked by episodic memory loss and impaired learning, followed by heterogenous changes in executive and visuospatial function [3]. The 2020 Lancet Commission on dementia prevention, intervention, and care identified a set of modifiable risk factors for late onset AD that include, but are not necessarily limited to, hypertension, smoking, obesity, physical inactivity, diabetes, alcohol, traumatic brain injury, and air pollution, and suggest that interventions that target such factors could prevent up to 40% of dementia prevalence [6][7][8]. Whereas age is the main risk factor for AD, more than 50 genetic risk loci are known, which include genes that regulate lipid homeostasis and inflammation, [9][10][11][12] with the strongest association being that with the ε4 allele of ApoE (ApoE4) [13][14][15]. In keeping with the genetic risk factors that predispose to AD, inflammation and oxidative stress feature prominently in such modifiable lifestyle factors.
2. Aβ/NFT Pathway
Despite the high prevalence of AD that has driven intense clinical and translational research efforts over the past 30 years, the complex pathophysiology remains poorly understood. The amyloid-beta (Aβ) and tau pathways proposed in the late 1980s and early 1990s have become hallmarks of AD [16]. The Aβ component involves the deposition of extracellular senile plaques of Aβ, products of cleavage of a larger amyloid precursor protein (APP) by β- and γ-secretases [16][17][18] (see Figure 1), that is naturally produced in the brain by neurons, vascular and blood cells, and astrocytes. APP is involved in multiple neuronal functions including neurite outgrowth and axonal guidance, the regulation of synaptic function and plasticity, early nervous system development, and neuroprotection [19]. Aβ aggregates progress from soluble oligomers and protofibrils to insoluble fibrils and plaques, all of which may be neurotoxic [20]. The second component involves accumulations of abnormally phosphorylated tau, a protein codified by the alternative splicing of the microtubule-associate protein tau (MAPT) gene that is enriched in axons of mature neurons, where it stabilizes the microtubules required for neuronal transport and structure. Hyperphosphorylated tau with a reduced affinity for tubulin becomes detached from microtubules and aggregates into paired helical filaments that spread and form cytoplasmic neurofibrillary tangles (NFTs) within neurons [20][21]. Neuropathological evidence indicates that increased levels of intracellular Aβ, parenchymal Aβ plaques, and NFTs are first seen in neurons of the entorhinal cortex (EC) [22][23][24].
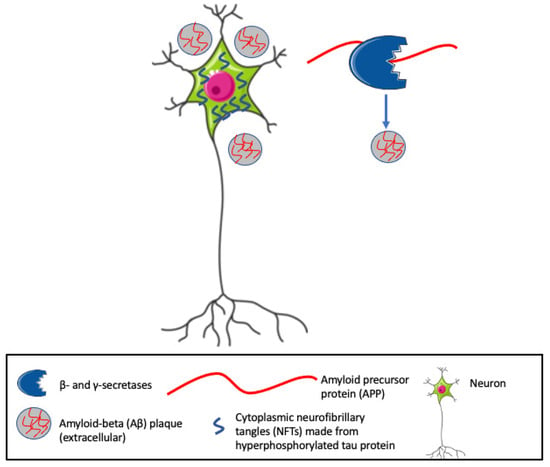
Figure 1. Hallmarks of Alzheimer’s Disease (AD), include cytoplasmic neurofibrillary tangles (NFTs) and extracellular amyloid beta (Aβ) plaques. β- and γ-secretases cleave the amyloid precursor protein (APP) into Aβ fragments that, along with hyperphosphorylated tau, generate neurotoxic aggregates and eventually insoluble plaques.
3. Etiology: Molecular Genetic and Epigenetic
Genetic evidence strongly supports the hypothesis that Aβ/tau initiates the early-onset form of AD. Large-scale genetic analyses of AD pedigrees identify highly penetrant mutations in APP, and genes encoding the subunits of the γ-secretase APP cleavage enzymes presenilin 1 and 2, as causal for dominantly inherited early-onset AD [25][26][27][28]. Mutations in the microtubule-associate protein tau (MAPT) gene that confer the abnormal production and aggregation of tau leading to NFT are also dominantly inherited and cause early-onset frontotemporal dementia [28]. The results are consistent with Aβ/NFT as diagnostic for AD. Whereas no causal genetic mutations are known for late-onset AD, that does not follow Mendelian inheritance, significant heritability is apparent, with evidence that the risk of AD may be as much as 80% dependent on heritable factors [9][10][11][29]. The expression of the ε4 allele of the ApoE gene (ApoE4) confers the highest risk for late-onset AD [10]. However, large-scale genome-wide association studies identified multiple additional critical genetic risk factors associated with more than 50 susceptibility gene loci including CLU, PICALM, CR1, BIN1, MS4A, CD2AP, CD33, EPHA1, and ABCA7 [30][31][32][33]. Many of these genes are linked directly to Aβ and tau homeostasis and include genes involved in inflammation and immune response pathways, cellular trafficking, lipid metabolism, and ubiquitination pathways [29]. Such studies revealed strong associations of variants in genes for immune receptors, TREM2 (triggering receptor expressed on myeloid cells 2) [34], and CD33 [35][36] with late AD. The results further corroborate the links between activated Aβ/tau, inflammation, and AD progression. By blocking the clearance of Aβ, vascular dysfunction is also implicated in AD progression and may be the mechanism whereby cerebral small vessel disease (CSVD) exacerbates AD [37][38][39][40]. Through its differential regulation of integrin-mediated signaling and neuroinflammation, OPN has long been known to have central roles in inflammatory CNS diseases including MS and AD, driving both neurodegenerative and protective pathways [41].
4. ApoE and LDLR
ApoE, the primary lipid and cholesterol transporters in the central nervous system, facilitate the transport of lipids by binding to LDLR and LRP1 [42]. ApoE is abundantly expressed in astrocytes, microglia, vascular mural cells, and choroid plexus cells, where it differentially modulates neuronal intracellular signaling pathways, including synaptic homeostasis, glucose metabolism, and cerebrovascular function [43]. Of the three ApoE isoforms expressed in the human brain, the presence of ApoE4 correlates with rapid AD onset and neurodegenerative decline and poses the highest genetic risk for AD, whereas ApoE2 is protective [13][14][15]. Amino acid substitutions on the N- and C-terminus of the proteins that, respectively, contain LDLR and lipid binding sites of ApoE proteins are responsible for the differences [44]. A large body of evidence indicates that ApoE4 binds more strongly to Aβ, promoting the aggregation, stabilization, and deposition of fibrillar plaques and reducing clearance, whereas ApoE2 binds weakly, reduces Aβ aggregates and NFTs, and enhances Aβ clearance by microglia [45][46].
Only non-lipidated ApoE binds Aβ plaque. Reducing the total ApoE levels in the brain or increasing its lipidation state markedly reduced the Aβ plaque burden and associated inflammatory phenotypes in mouse AD models [46][47][48][49][50]. Such restricted control of the Aβ/NFT burden to non-lipidated ApoE is also consistent with the established roles of the LDLR and LRP1 in regulating ApoE and Aβ plaque.
5. Inflammation
Neuronal microgliosis (migration of local macrophages, astrocytes, and microglia) constitutes the principal inflammatory response to Aβ plaque deposition and correlates closely with the onset of brain cell damage and cognitive decline [51][52]. However, mouse models and patient studies also implicate significant peripheral immune cell infiltration of the brain parenchyma during different stages of AD [53]. Systemic inflammation driven by Aβ egress into the circulation via two-way transport systems such as the LDLR associated factors that circumvent the blood–brain barrier (BBB) [54][55] may contribute to AD progression by interfering with the clearance of Aβ by microglia, triggering tau hyperphosphorylation and the spread of NFT, as well as by promoting BBB breakdown [56][57][58][59][60]. Systemic myeloid cells and inflammatory cytokines can access the brain via humoral and vascular signaling pathways and confer both destructive and reparative actions [60][61][62]. However, during active AD inflammation, Aß plaques and regions of the brain with high tau accumulation are frequently found surrounded by CD11b+, Iba1+, and CD45+ immune cells that originate mostly if not entirely from microglia [63][64][65][66]. The primary function of such microglial migration is to repair and clear Aß plaque through phagocytosis and autophagy, but in the context of late-stage symptomatic AD, effective clearance fails. The reasons for failure are not understood but presumably involve overwhelmed clearance systems, due in part to the relatively late appearance of phagocytic/anti-inflammatory microglia (see below), inhibitory cytokines, and adaptive responses to the long latent period of Aβ/NFT evolution, and the continued presence of toxic AβOs [66][67].
Activated microglia can be neurotrophic or neurotoxic depending on the activating stimuli, immune milieu, and surrounding environment [68][69][70]. M1 or inflammatory microglia are usually activated via Toll-like receptors and γ-interferon signaling, producing pro-inflammatory cytokines such as interleukins (IL-1β) and tumor necrosis factor alpha (TNF-a) and chemokines, and expressing NADPH oxidase and matrix metalloproteinases [62][65]. Anti-inflammatory M2 microglia are neuroprotective, secrete growth factors, and promote the release of anti-inflammatory cytokines (reviewed in [3][71]). OPN interacts with both M1- and M2-activated microglia, showing synergistic detrimental and protective functions, respectively [41][72].
Microglia are increasingly understood to play flexible and sometimes opposing roles in AD pathogenesis by eliminating toxic Aβ aggregates, enhancing neuronal plasticity and conferring anti-inflammatory signals, or by producing proinflammatory cytokines, ROS, and neurotoxicity [73]. Microglia express cell-surface receptors that differentially activate innate immune responses and mediate protective and destructive responses to Aβ aggregates.
6. Osteopontin
OPN is a negatively charged acidic phosphoprotein that circulates as a pro-inflammatory cytokine [74]. Variously referred to as early T-cell activation protein (ETA-1), bone sialoprotein (BSP-1), and secreted phosphoprotein (SPP-1), OPN is multifunctional, with different functional domains exposed through thrombin or metalloprotease cleavage [3][41] (see Figure 2). Thrombin cleavage generates an N-terminal OPN fragment that binds integrins and mediates cell adhesion and migration, bone marrow immune responses, and inflammation [3]. OPN is found in the bone matrix, kidney, placenta, and blood vessels and contributes to multiple physiologic and pathologic processes, including bone mineralization, oxidative stress, remyelination, inflammation, immunity, and wound healing [74][75]. OPN, secreted by T cells and tissue-resident macrophages, has been shown to regulate macrophage functions, including migration, reparative and degenerative phagocytosis, and chemotaxis in different peripheral tissues [76][77][78].
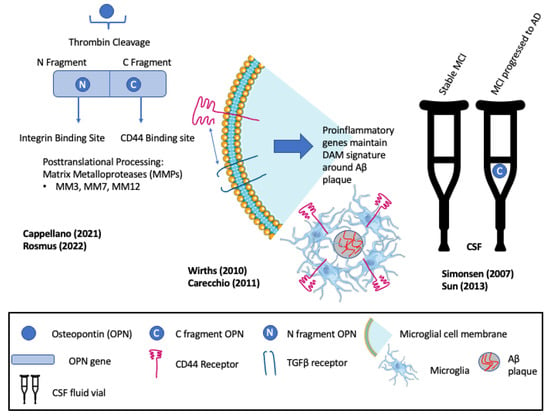
Figure 2. Multifunctionality of osteopontin (OPN) and its roles in recruiting and maintaining disease-associated microglia (DAM). Elevated levels of the C-fragment of OPN in cerebrospinal fluid (CSF) mark the progression of patients with mild cognitive impairment (MCI) to clinical Alzheimer’s disease (AD). Rosmus et al., Wirths et al., Cappellano et al., Sun et al., Simonsen et al., Carecchio et al. [3][21][41][74][79][80].
Early evidence for roles of OPN in neurodegenerative disease reported an association with severe intracranial calcification, wherein calcification within the basal ganglia and cerebellum, including type 3 capillary calcospherites, express elevated OPN [75]. Similarly, OPN is found to be associated with the TGFβ signaling factor phosphor-SMAD2/3 and collagen-1 in calcified vessel walls of the cerebral cortex of patients with cerebral amyloid angiopathy [81]. Subsequently, increased OPN expression was described in the pyramidal neurons of the CA1 region of the hippocampus associated with Aβ plaque of symptomatic AD patients [82]. Proteomic analyses of CSF from patients with stable MCI versus MCI with AD identified the phosphorylated C-terminal thrombin cleavage fragment of OPN as a biomarker of MCI that progressed to AD [79]. Positive correlations between the levels of the OPN C-terminal fragment in the CSF and elevated inflammatory markers suggested a mechanistic link between OPN and increased inflammation and gliosis as MCI progressed to AD [79] (see Figure 2).
Elevated OPN expression in the astrocytes and microglia of ADtg mice was shown to correlate with similarly elevated levels of inflammation and oxidative stress markers [21]. OPN, GFAP, Cathepsin D, Toll-like receptors (TLRs), and TGFβ-1 correlated with the progressive age-dependent loss of CA1 pyramidal neurons, axons, and quantitative deficits in cognitive and motor performance. The results identified OPN as a marker of astrocytes and microglial activation and inflammation [21]. Consistent with this, and in agreement with the proteomic analyses described above [79], elevated OPN levels in CSF are also increased in patients with mild cognitive impairment (MCI) that progress to AD, but not in patients with stable MCI [74] (see Figure 2).
In support of pro-inflammatory, neurodegenerative roles of OPN, represented in Figure 3 and Figure 4, one study highlighted that the antibody-mediated activation of the TREM2 receptor in ADtg mice displayed enhanced survival and proliferation of microglia and ameliorated Aβ-induced pathology and the downregulation of OPN [83][84] (See Figure 3). TREM2 is a receptor for microglial lipids and a genetic risk factor for AD. The results identified OPN as a marker of inflammation that correlates positively with neurodegeneration and negatively with protective microglia migrations to sites of Aβ plaque. Consistent with this, De Schepper et al. report that OPN secreted by hippocampal perivascular macrophages (PVM), the primary responders to toxic agents and pathogens that cross the BBB, transformed microglia into phagocytic cells that engulf synapses in ADtg mice and possibly AD patient tissues [85]. OPN was found to upregulate multiple phagocytic markers including C1qa, Grn, and Ctsb in microglia associated with Aβ oligomers (see Figure 4).
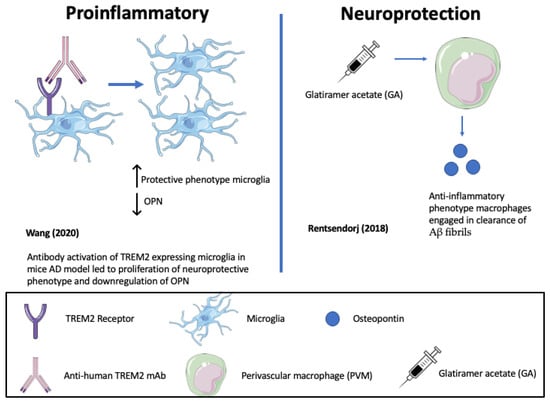
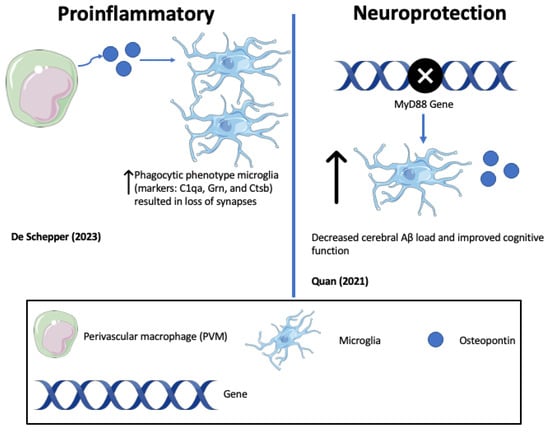
Figure 4. Opposing roles of osteopontin (OPN) in driving Alzheimer’s disease (AD) pathology (De Schepper (2023) [85]; Quan (2021) [86]).
Conversely, as highlighted in Figure 3 and Figure 4, in other mouse ADtg models and in human AD brains, elevated OPN expression correlated with protective, anti-inflammatory phenotypes and accelerated Aβ clearance [61]. When ADtg mice were injected with glatiramer acetate (GA), an immune-modulating medication used to treat MS patients, the OPN expression associated with monocytes and macrophages of systemic origin engaged in Aβ clearance and tissue repair and was found in areas of high Aβ plaque. Infiltrating OPN-expressing cells included subpopulations of CD115+CD11b+Ly6Chigh monocytes and CD11b+Ly6C+CD45high monocyte/macrophages. Similar effects were achieved by delivering peripheral blood enriched with bone marrow (BM)-derived CD115+ monocytes. Correlation matrix analyses indicated a strong linear correlation between cerebral OPN levels and macrophage infiltration, and an inverse relation between OPN and Aβ plaque burden. In vitro studies corroborated the findings, showing that GA directly upregulates OPN expression in BM-derived macrophages and promotes a phenotypic shift to phagocytic, anti-inflammatory effect with an increased uptake of Aβ fibrils [61] (see Figure 3).
It is noteworthy that OPN C- and N-terminal fragments were previously shown to predict the presence of AD conversion in MCI patients [74][79]. Taken together, these studies suggest that the microglial expression of OPN is associated with pro-inflammatory, neurodegenerative pathways during AD, whereas OPN expressing systemic monocytes confers protective/reparative functions. Further, extracellular OPN can transform subpopulations of microglia to a phagocytic pro-inflammatory subset that engulfs and destroys synapses while BM-derived monocytes are converted to phagocytic anti-inflammatory states that engulf and remove Aβ. Therefore, the actions of OPN depend on the context, cell types, stage of disease, and prevailing biochemical interactions. Protection from AD progression in OPN-ablated ADtg mice suggests a net pathological/neurodegenerative effect. These complex properties with apparently opposing functions in different cells at disease stages make OPN a challenging target for AD therapy that will require physical and temporally restrictive exposure to modulating drugs, and/or interventions that target downstream signaling molecules.
In another approach to unravel the role of OPN in microglial activation, Qiu et al. identified a subset of CD11+ microglia that are the sole source of OPN in ADtg mice [87] (see Figure 5). The genetic deletion of OPN in ADtg mice markedly reduced proinflammatory microglia, Aβ plaques, and dystrophic neurites, while conserving cognitive function. OPN production in the periplaque areas was found to differentiate DAM into two distinct subpopulations, including a protective CD11c+OPN− subset with an increased expression of activated lysosomal markers that robustly takes up Aβ in a noninflammatory manner, and a pathogenic CD11c+OPN+ subset that produces proinflammatory cytokines including TNF-α, expresses reduced periplaque levels of TREM2, and does not ingest Aβ fibrils. Both subsets of microglia localized around plaques. The authors went on to demonstrate a threefold increase in CD11c+ OPN+ microglia in brain sections from AD patients compared with normal controls or MCI patients.
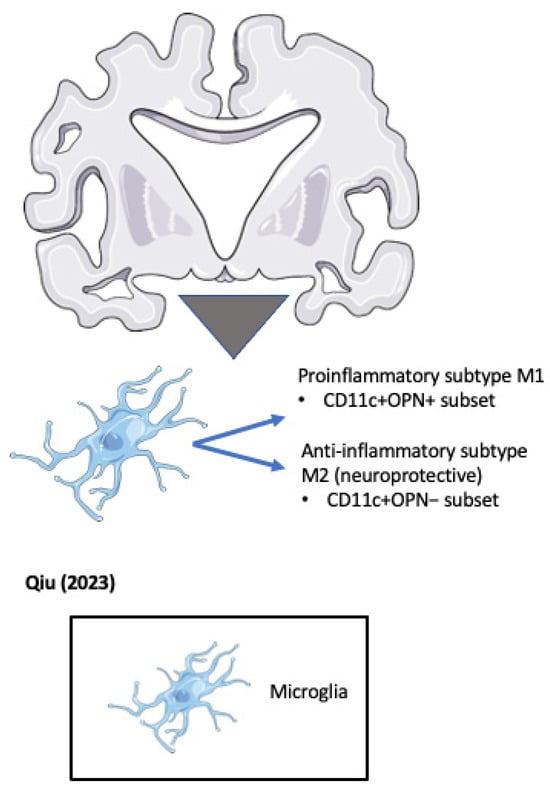
Figure 5. OPN positive and negative microglial subsets accumulate around pathogenic plaques in the Alzheimer’s disease (AD) brain (Qiu et al. 2023) [87].
7. Conclusions
The presence of multiple isoforms of microglia with protective and disease-associated phenotypes, and the property of microglial subtypes to switch phenotypes depending on environmental/cellular cues, suggest that OPN directs pro-inflammatory or anti-inflammatory signals depending on the myeloid and/or microglial cells with which it interacts, and with the disease stage, and context of the targeted cells. The neuroprotective actions of OPN-associated bone marrow leukocytes described by Rentsendorj et al. [61] associated with advanced Aβ plaque deposits suggest that OPN confers neuroprotective and/or reparative functions via systemic immunity at late-stage AD when neuronal damage is established and ongoing. The work is consistent with previous reports that macrophages derived from the BM phagocytose Aβ plaques more efficiently than resident microglia [88][89][90]. However, OPN-mediated neurotoxicity by multiple pathways involving microglia appears to predominate via pathways that include the enhanced formation and toxicity of Aβ plaques and NFTs, increased activities of APP cleavage enzymes, interference with Aβ clearance, and the delivery of proinflammatory cytokines. OPN can promote toxic tau accumulation and NFT fibers and contribute to disrupting the BBB, allowing the access of additional inflammatory mediators, immune cells, or pathogens to enter the brain [91]. Recently, the interrogation of human iPSC-derived microglia by CRISPR-based functional genomics identified an SPP1+ microglial subtype with a neurodegenerative disease-specific state that was targeted and neutralized by PLX3397, a selective inhibitor of colony-stimulating factor-1 [70]. PLX3397 was previously shown to be protective in neurodegenerative mouse models [92][93]. In another cell-based therapeutic approach, human neural-crest-derived turbinate stem cells were shown to neutralize Aβ neurotoxicity by suppressing OPN expression in a human cerebral organoid model of AD and in ADtg mice [94]. Thus OPN/Spp1 is considered to be a negative DAM-associated gene and AD therapeutic target. However, because of possible OPN-directed protection mediated by systemic immunity, the outcome of OPN blockade in late-stage AD cannot be predicted.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11123232
References
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569.
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789.
- Rosmus, D.D.; Lange, C.; Ludwig, F.; Ajami, B.; Wieghofer, P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines 2022, 10, 840.
- Chai, Y.L.; Chong, J.R.; Raquib, A.R.; Xu, X.; Hilal, S.; Venketasubramanian, N.; Tan, B.Y.; Kumar, A.P.; Sethi, G.; Chen, C.P.; et al. Plasma osteopontin as a biomarker of Alzheimer’s disease and vascular cognitive impairment. Sci. Rep. 2021, 11, 4010.
- Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125.
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37.
- Waite, L.M. Treatment for Alzheimer’s disease: Has anything changed? Aust. Prescr. 2015, 38, 60–63.
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446.
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174.
- Avramopoulos, D. Genetics of Alzheimer’s disease: Recent advances. Genome Med. 2009, 1, 34.
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444.
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. J. Am. Med. Assoc. 2023, 330, 512–527.
- Poirier, J.; Davignon, J.; Bouthillier, D.; Kogan, S.; Bertrand, P.; Gauthier, S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 1993, 342, 697–699.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923.
- Caselli, R.J.; Dueck, A.C.; Osborne, D.; Sabbagh, M.N.; Connor, D.J.; Ahern, G.L.; Baxter, L.C.; Rapcsak, S.Z.; Shi, J.; Woodruff, B.K.; et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl. J. Med. 2009, 361, 255–263.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552–7556.
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51.
- Chau, D.D.; Ng, L.L.; Zhai, Y.; Lau, K.F. Amyloid precursor protein and its interacting proteins in neurodevelopment. Biochem. Soc. Trans. 2023, 51, 1647–1659.
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265.
- Wirths, O.; Breyhan, H.; Marcello, A.; Cotel, M.C.; Brück, W.; Bayer, T.A. Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 747–757.
- Ridler, C. Tau seeding starts early in the entorhinal cortex. Nat. Rev. Neurol. 2018, 14, 380.
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969.
- Kobro-Flatmoen, A.; Lagartos-Donate, M.J.; Aman, Y.; Edison, P.; Witter, M.P.; Fang, E.F. Re-emphasizing early Alzheimer’s disease pathology starting in select entorhinal neurons, with a special focus on mitophagy. Ageing Res. Rev. 2021, 67, 101307.
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977.
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778.
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706.
- Pagnon de la Vega, M.; Näslund, C.; Brundin, R.; Lannfelt, L.; Löwenmark, M.; Kilander, L.; Ingelsson, M.; Giedraitis, V. Mutation analysis of disease causing genes in patients with early onset or familial forms of Alzheimer’s disease and frontotemporal dementia. BMC Genom. 2022, 23, 99.
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322.
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441.
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435.
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099.
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127.
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850.
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643.
- Kim, S.E.; Kim, H.J.; Jang, H.; Weiner, M.W.; DeCarli, C.; Na, D.L.; Seo, S.W. Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. Int. J. Mol. Sci. 2022, 23, 10490.
- Kim, H.J.; Yang, J.J.; Kwon, H.; Kim, C.; Lee, J.M.; Chun, P.; Kim, Y.J.; Jung, N.Y.; Chin, J.; Kim, S.; et al. Relative impact of amyloid-β, lacunes, and downstream imaging markers on cognitive trajectories. Brain 2016, 139, 2516–2527.
- Jang, H.; Park, J.Y.; Jang, Y.K.; Kim, H.J.; Lee, J.S.; Na, D.L.; Noh, Y.; Lockhart, S.N.; Seong, J.K.; Seo, S.W. Distinct amyloid distribution patterns in amyloid positive subcortical vascular cognitive impairment. Sci. Rep. 2018, 8, 16178.
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jäger, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain 2017, 140, 1107–1116.
- Cappellano, G.; Vecchio, D.; Magistrelli, L.; Clemente, N.; Raineri, D.; Barbero Mazzucca, C.; Virgilio, E.; Dianzani, U.; Chiocchetti, A.; Comi, C. The Yin-Yang of osteopontin in nervous system diseases: Damage versus repair. Neural Regen. Res. 2021, 16, 1131–1137.
- Kim, J.; Castellano, J.M.; Jiang, H.; Basak, J.M.; Parsadanian, M.; Pham, V.; Mason, S.M.; Paul, S.M.; Holtzman, D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 2009, 64, 632–644.
- Nyarko, J.N.K.; Quartey, M.O.; Pennington, P.R.; Heistad, R.M.; Dea, D.; Poirier, J.; Baker, G.B.; Mousseau, D.D. Profiles of β-Amyloid Peptides and Key Secretases in Brain Autopsy Samples Differ with Sex and APOE ε4 Status: Impact for Risk and Progression of Alzheimer Disease. Neuroscience 2018, 373, 20–36.
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344.
- Fitz, N.F.; Cronican, A.A.; Saleem, M.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 2012, 32, 13125–13136.
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63.
- Huynh, T.V.; Davis, A.A.; Ulrich, J.D.; Holtzman, D.M. Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 2017, 58, 824–836.
- Farfel, J.M.; Yu, L.; De Jager, P.L.; Schneider, J.A.; Bennett, D.A. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 2016, 37, 19–25.
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155.
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 2021, 13, eabd7522.
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242.
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081.
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener. Dis. 2018, 18, 49–56.
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987.
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. 2014, 12, 206.
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615.
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6.
- Tao, Q.; Ang, T.F.A.; DeCarli, C.; Auerbach, S.H.; Devine, S.; Stein, T.D.; Zhang, X.; Massaro, J.; Au, R.; Qiu, W.Q. Association of Chronic Low-grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw. Open 2018, 1, e183597.
- Yeung, C.H.C.; Schooling, C.M. Systemic inflammatory regulators and risk of Alzheimer’s disease: A bidirectional Mendelian-randomization study. Int. J. Epidemiol. 2021, 50, 829–840.
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2021, 12, 796867.
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180.
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74.
- Shi, Y.; Andhey, P.S.; Ising, C.; Wang, K.; Snipes, L.L.; Boyer, K.; Lawson, S.; Yamada, K.; Qin, W.; Manis, M.; et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 2021, 109, 2413–2426.e7.
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183.e8.
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10.
- Shukla, A.K.; McIntyre, L.L.; Marsh, S.E.; Schneider, C.A.; Hoover, E.M.; Walsh, C.M.; Lodoen, M.B.; Blurton-Jones, M.; Inlay, M.A. CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia 2019, 67, 844–856.
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610.
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594.
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753.
- Dräger, N.M.; Sattler, S.M.; Huang, C.T.; Teter, O.M.; Leng, K.; Hashemi, S.H.; Hong, J.; Aviles, G.; Clelland, C.D.; Zhan, L.; et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat. Neurosci. 2022, 25, 1149–1162.
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front. Neurol. 2023, 14, 1103416.
- Rabenstein, M.; Vay, S.U.; Flitsch, L.J.; Fink, G.R.; Schroeter, M.; Rueger, M.A. Osteopontin directly modulates cytokine expression of primary microglia and increases their survival. J. Neuroimmunol. 2016, 299, 130–138.
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724.
- Sun, Y.; Yin, X.S.; Guo, H.; Han, R.K.; He, R.D.; Chi, L.J. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediat. Inflamm. 2013, 2013, 615745.
- Fujita, D.; Terada, S.; Ishizu, H.; Yokota, O.; Nakashima, H.; Ishihara, T.; Kuroda, S. Immunohistochemical examination on intracranial calcification in neurodegenerative diseases. Acta Neuropathol. 2003, 105, 259–264.
- Rittling, S.R. Osteopontin in macrophage function. Expert. Rev. Mol. Med. 2011, 13, e15.
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10-STAT3-Galectin-3 Axis Is Essential for Osteopontin-Producing Reparative Macrophage Polarization After Myocardial Infarction. Circulation 2018, 138, 2021–2035.
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e14.
- Simonsen, A.H.; McGuire, J.; Hansson, O.; Zetterberg, H.; Podust, V.N.; Davies, H.A.; Waldemar, G.; Minthon, L.; Blennow, K. Novel Panel of Cerebrospinal Fluid Biomarkers for the Prediction of Progression to Alzheimer Dementia in Patients with Mild Cognitive Impairment. Arch. Neurol. 2007, 64, 366–370.
- Carecchio, M.; Comi, C. The role of osteopontin in neurodegenerative diseases. J. Alzheimers Dis. 2011, 25, 179–185.
- Grand Moursel, L.; van der Graaf, L.M.; Bulk, M.; van Roon-Mom, W.M.C.; van der Weerd, L. Osteopontin and phospho-SMAD2/3 are associated with calcification of vessels in D-CAA, an hereditary cerebral amyloid angiopathy. Brain Pathol. 2019, 29, 793–802.
- Wung, J.K.; Perry, G.; Kowalski, A.; Harris, P.L.; Bishop, G.M.; Trivedi, M.A.; Johnson, S.C.; Smith, M.A.; Denhardt, D.T.; Atwood, C.S. Increased expression of the remodeling- and tumorigenic-associated factor osteopontin in pyramidal neurons of the Alzheimer’s disease brain. Curr. Alzheimer Res. 2007, 4, 67–72.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116.
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785.
- De Schepper, S.; Ge, J.Z.; Crowley, G.; Ferreira, L.S.S.; Garceau, D.; Toomey, C.E.; Sokolova, D.; Rueda-Carrasco, J.; Shin, S.H.; Kim, J.S.; et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 406–415.
- Quan, W.; Luo, Q.; Hao, W.; Tomic, I.; Furihata, T.; Schulz-Schäffer, W.; Menger, M.D.; Fassbender, K.; Liu, Y. Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. Glia 2021, 69, 1987–2005.
- Qiu, Y.; Shen, X.; Ravid, O.; Atrakchi, D.; Rand, D.; Wight, A.E.; Kim, H.J.; Liraz-Zaltsman, S.; Cooper, I.; Schnaider Beeri, M.; et al. Definition of the contribution of an Osteopontin-producing CD11c+ microglial subset to Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2218915120.
- Koronyo-Hamaoui, M.; Ko, M.K.; Koronyo, Y.; Azoulay, D.; Seksenyan, A.; Kunis, G.; Pham, M.; Bakhsheshian, J.; Rogeri, P.; Black, K.L.; et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem. 2009, 111, 1409–1424.
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422.
- Bernstein, K.E.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.T.; Yu, J.J.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012.
- Ueno, M.; Chiba, Y.; Matsumoto, K.; Murakami, R.; Fujihara, R.; Kawauchi, M.; Miyanaka, H.; Nakagawa, T. Blood-brain barrier damage in vascular dementia. Neuropathology 2016, 36, 115–124.
- Zhan, L.; Fan, L.; Kodama, L.; Sohn, P.D.; Wong, M.Y.; Mousa, G.A.; Zhou, Y.; Li, Y.; Gan, L. A MAC2-positive progenitor-like microglial population is resistant to CSF1R inhibition in adult mouse brain. eLife 2020, 9, e51796.
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.H.; Sayed, F.; et al. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol. 2019, 17, e3000134.
- Lim, J.Y.; Lee, J.E.; Park, S.A.; Park, S.I.; Yon, J.M.; Park, J.A.; Jeun, S.S.; Kim, S.J.; Lee, H.J.; Kim, S.W.; et al. Protective Effect of Human-Neural-Crest-Derived Nasal Turbinate Stem Cells against Amyloid-β Neurotoxicity through Inhibition of Osteopontin in a Human Cerebral Organoid Model of Alzheimer’s Disease. Cells 2022, 11, 1029.
This entry is offline, you can click here to edit this entry!