A standardized assessment of Tumor Mutational Burden (TMB) poses challenges across diverse tumor histologies, treatment modalities, and testing platforms, requiring careful consideration to ensure consistency and reproducibility. Despite clinical trials demonstrating favorable responses to immune checkpoint inhibitors (ICIs), not all patients with elevated TMB exhibit benefits, and certain tumors with a normal TMB may respond to ICIs. Therefore, a comprehensive understanding of the intricate interplay between TMB and the tumor microenvironment, as well as genomic features, is crucial to refine its predictive value. Bioinformatics advancements hold potential to improve the precision and cost-effectiveness of TMB assessments, addressing existing challenges.
1. Background
Tumor Mutational Burden (TMB) has emerged as a promising genomic biomarker of response to immune checkpoint inhibitors that was pioneered by Rizvi and Chan [
1]. TMB is defined as the total number of mutations within a tumor genome (mutations per megabase of genome or mut/Mb) and serves as a measure of the potential neoantigen load that may elicit an anti-tumor immune response. This is based upon the type of mutations, a frequency of threshold in the context of clinical activity, the type of cancer cohort, and the sequencing methods used (whole-exome sequencing (WES), whole-genome sequencing (WGS), targeted panel-based next-generation sequencing (NGS)) [
2].
TMB molecular signatures are varied, and they arise from a combination of both exogenous and endogenous factors, which represent environmental factors that affect mutagenesis rates and mutations due to random errors in deoxyribonucleic acid (DNA) replication, respectively [
3]. This molecular signature has been shown to be predictive of response to immune checkpoint inhibitors (ICIs) in several clinical trial settings. Similarly, hypermutated tumors vary in their causative factors, for example, UV (ultraviolet) light for skin cancer and smoking in non-small cell lung cancer (NSCLC), as well as somatic mutations. In addition, there is a huge variability in mutational load across cancers, with melanoma and NSCLC frequently showing a high TMB. Thus, defining a median range across several cancer types is not practical and other approaches might serve as useful predictive biomarkers of response [
4]. Despite these adjustments, cancers such as renal cell cancer (RCC) may not have a high TMB but respond well to ICI.
High TMB is common among several cancers, with an incidence of TMB ≥ 10 mut/Mb of 14.7% based on the TSO500 (TruSight Oncology 500, Illumina, San Diego, CA, USA) panel assay in one study and a pooled overall prevalence of 14% on panel-based or WES assays in solid tumors [
5,
6]. Among less common tumors, including but not limited to biliary tract cancers, small cell lung cancer, and mesothelioma, the prevalence of TMB-H (≥10 mut/Mb on FoundationOne®CDx (F1CDx) panels) was noted to be 12.8% [
7].
2. Challenges of Using TMB as Biomarker of Response to ICI
Whole-exome sequencing (WES) encompasses 32 Mb of the coding region, the entire set of 22,000 genes, which constitutes roughly 1% of the genome. WES, the gold standard for TMB calculation, measures the number of somatic alterations, whereas the FDA-authorized surrogate panel assays including the Memorial Sloan-Kettering-Integrated Mutation Profiling of Actionable Cancer Targets or MSK-IMPACT (468 genes) and F1CDx assay (324 genes) measure the density of somatic alterations, including target footprints of approximately 1.14 Mb and 0.8 Mb of the coding regions, respectively [
12,
13].
Keynote-158, the pivotal trial leading to the tissue-agnostic approval of pembrolizumab, evaluated tissue TMB (tTMB) in FFPE (Formalin-Fixed Paraffin-Embedded) tumor samples through the utilization of the F1CDx assay with a predefined criterion for classifying high tTMB as the presence of a minimum of 10 mut/Mb. The trial showed an objective response rate (ORR) of 29% in the tTMB-high versus 6% in the non-tTMB-high groups when treated with pembrolizumab at 200 mg IV (intravenously) every 3 weeks until unacceptable toxicity or disease progression [
15].
The overall survival (OS) and progression-free survival (PFS) were secondary outcomes in the trial, and a mOS (median OS) benefit was not seen (11.5 months in t-TMB-high versus 12.8 months in non-tTMB-high group). The trial was certainly limited in the types of cancers (it included nine different cancer types) and patient numbers, including cancers with relative resistance to immunotherapy. Fourteen percent of patients assessed for efficacy in the TMB-high group also had Microsatellite Instability-High (MSI-H); however, after excluding patients with missing or MSI-H status, the ORR was 28%.
A retrospective analysis of several keynote trials (Keynote-001, Keynote-002, Keynote-010, Keynote-012, Keynote-028, Keynote-045, Keynote-055, Keynote-059, Keynote-061, Keynote-086, Keynote-100) demonstrated improvement in ORR among patients treated with ICI, with tTMB ≥ 175 mutations/exome compared to TMB < 175 mutations/exome (31.4% vs. 9.5%, respectively). Additionally, an analysis of patients in Keynote-010, Keynote-045, and Keynote-061 showed improvements in PFS and OS compared to chemotherapy [
18]. A more recent retrospective analysis also showed that patients in Keynote-042 derived PFS and OS benefits if their tTMB was ≥ 175 mutations/exome compared to tTMB < 175 mutations/exome [
19]. Multiple other systematic reviews and metanalyses have indicated a strong association between high TMB and enhanced efficacy, with several studies also highlighting improved survival outcomes across cancers and in specific cancer types [
20,
21,
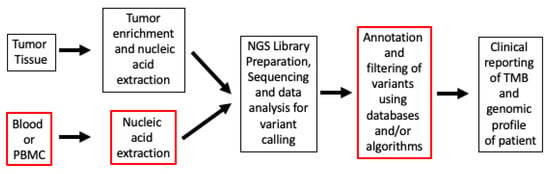
22]. When it comes to evaluating TMB, inherent differences exist in testing workflow methods (
Figure 1).
Figure 1. TMB workflow from tissue specimens to clinical report. The boxes with red outlines are steps that are substantially different in labs and workflows. Abbreviations: NGS, next-generation sequencing; PBMC, peripheral blood mononuclear cells; TMB, Tumor Mutational Burden.
While WES and MSK-IMPACT use similar filtering strategies of using non-synonymous mutations for calculating TMB, tumor-only sequencing panels (like F1CDx) also include short indels in introns and synonymous mutations in the panel testing for TMB. And, considering that neoantigens are formed by non-synonymous mutations only, the synonymous mutations are included in TMB calculation to reduce sampling noise and increase the robustness of TMB scoring methods [
23]. In some cases, the use of synonymous mutations in conjunction with a statistical framework substantially increased the concordance of TMB values with a WES-based reference method [
24].
Additional differences in the panel content among the commercial panel assays may give rise to inter-assay variations. For example, in focused panels, there may be higher detection rates of pathogenic driver mutations compared to the background mutation rate in the tumor. This may lead to higher levels of variability at low TMB values [
26]. Therefore, it may be important to calibrate the thresholds for each panel in addition to generating an adjusted score (e.g., “mutational load”) by taking into account the variability within tumor types [
31]. This may help in better interpretation of the TMB scores. Many of these panel-adjusted scores used in determining TMB also show a good correlation to TMB scores generated from WES and WGS [
32], whereas the TMB calculation from WES and WGS seems to be highly concordant, although the data analysis pipeline for WES is less intensive and less costly [
32,
33].
Several factors impact the TMB calculation among different panel tests. The first of these is the size of the panel variation, such as 0.8 Mb in F1CDx versus 1.94 Mb for the TSO500 panel. Studies have shown that a smaller panel size may produce more misclassifications of TMB calculation compared to a larger panel size. This includes variation in thresholds among smaller compared to larger panel sizes. In particular, a smaller panel size is less precise in distinguishing between hypermutated from non-hypermutated cancers [
34]. Smaller panel sizes also tend to overestimate TMB values whereas panel tests may overestimate TMB compared to WES [
35].
Inter- and intra-tumor heterogeneity can produce imprecise TMB measurement [
37]. TMB from metastatic sites could be higher than that from primary sites due to varying clonal heterogeneity, and yet, this disparity may not necessarily influence the survival benefit derived from ICI therapy [
38].
Tumor-targeting cytotoxic T lymphocyte variation in the cellular neighborhoods (CNs) is dynamic and impacts response to ICI therapy, with HLA-1 expression downregulation predicting poor outcomes with ICI in colorectal cancer patients [
41].
Another challenge lies in the influence of tumor purity, such as that related to tissue sampling, where the low tumor cellularity could result in falsely low TMB measurement [
42]. The exclusion of germline alterations also varies across different panels.
The composition of the panel test, with the variable selection of genomic alterations, can also produce variability in TMB calculation [
36]. After accounting for artefacts and germline variation, a panel test comparison shows a good correlation with the inclusion of synonymous and coding non-synonymous alterations [
44].
Overestimation in TMB can also occur by including mutations with VAF (variant allele frequency) only above a certain threshold [
45]. Hence, the specific context of the underlying mutation (synonymous, non-synonymous, or Indels) and whether it occurs in the coding or non-coding regions might contribute to minor variation in TMB calculation across different panels.
3. Factors in Tumor Microenvironment (TME), TMB, and Response to ICI
The tumor microenvironment (TME) constitutes a diverse ecosystem, and to harness the entire TME for improved immunotherapies, it is crucial to recognize that multiple immune subsets play a role in shaping the variability in immune response [
48,
49]. Thus, these anti-tumor immune responses are complex and involve several factors driving the cancer immunity cycle that promote or suppress anti-tumor immunity [
50]. Simply relying on a single biomarker such as TMB to explain the response to ICIs may not capture the intricate interplay of sensitivity and resistance mechanisms underlying the use of these therapies [
51].
It is also known that beyond tumor histology, there are several other mechanisms that can impact response to ICIs, such as cellular signaling, checkpoint signaling pathways, immune cell activity, variability in HLA expression and TCR repertoire, the gut microbiome, and oncogenic signaling pathways indirectly associated with response to ICIs [
52,
53].
Exploration in this field is an active area of clinical and translational research. This involves combining PD-1/PD-L1 (anti-programmed death receptor-1/anti-programmed death ligand-1) inhibitors with other immune-modulating or targeted agents, depending on the stromal environment of the tumor, for example in hot versus cold tumor microenvironments.
A higher HLA class II expression has been shown to be associated with positive tumor responses through correlative analysis in CheckMate 064 and CheckMate 069 [
56]. Similarly, heterozygosity in HLA-1 is associated with better response to ICIs [
57].
Immunologically cold tumors have lower response rates to ICIs. However, TMB-H is not always correlated with CD8+ tumor-infiltrating T-cells, since a portion of CD8+ T-cells are bystanders and recognize antigens unrelated to tumors [
58,
59].
A post hoc pan-cancer analysis using MSK-IMPACT for TMB scores showed that the OS among patients treated with ICIs was associated with sets of genomic alterations in TMB-low versus TMB-high tumors. In particular, hypermutation (TMB ≥ 100 mut/mb) is associated with certain genomic signatures including
POLE/
POLD1, dMMR (deficient mismatch repair), the activation of AID/APOBEC (activation-induced cytidine deaminase/apolipoprotein B mRNA (messenger ribonucleic acid) enzyme catalytic polypeptide-like), and the three clock-like mutational processes (SBS1, SBS5) [
26].
Somatic mutations have the potential to generate neoantigens, and the resulting cancer-specific genomic signatures can vary across cancer types [
61].
The impact of driver genes on TMB is noteworthy in NSCLC, wherein high TMB may be linked to reduced survival in
EGFR mutated cancers [
64]. Specific mutations, including
CDH1 (cadherin-1),
RAD50, and
MSH2 (muts Homolog 2), have been associated with high TMB in head and neck squamous cell cancers [
65]. Certain mutations linked to responses to ICIs have been observed [
66].
4. Efficacy and Real-World Data on TMB Testing
Several clinical trials have demonstrated the clinical utility of TMB as a predictive biomarker of response to single-agent and combination immunotherapy [
71]. In general, there is substantial real-world evidence indicating a response and enhanced survival in cases of TMB-H tumors, with varying definitions, but primarily in TMB thresholds ≥ 10 mut/Mb. The analysis of large clinico-genomic databases to assess real-world OS analyses of TMB has shown the benefit of high TMB across 24 cancer types compared to a low TMB [
72]. Another retrospective analysis of patients with MSI-H and/or TMB-H (≥20 mut/Mb on F1CDx) among 27 different cancer types showed better PFS outcomes with immunotherapy in patients who had previously received chemotherapy [
73]. A study in NSCLC demonstrated an increase in real-world OS as TMB scores increased from <10 to 10–19 and ≥20 mut/Mb (10.1, 11.8, and 26.9 months, respectively) [
74].
5. Conclusions
In conclusion, while not a perfect biomarker, with advancements in TMB measurement, standardization initiatives, enhanced testing protocols, TMB characterization in diverse cancer types, its amalgamation with other biomarkers of response to ICIs, dynamic monitoring through bTMB, and the rigorous validation of improved testing methodologies, among various other factors, TMB has the potential for enhanced practical utility in the real-world clinical setting.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15245841