Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly termed nonalcoholic fatty liver disease (NAFLD), is a widespread global health concern that affects around 25% of the global population. Its influence is expanding, and it is anticipated to overtake alcohol as the leading cause of liver failure and liver-related death worldwide. Mitochondrial dysfunction is strongly linked to the development of MASLD.
- liver
- mitochondrial dysfunction
- FAO
- MASLD
- thyroid hormone
- thyroid hormone receptor
1. Introduction
TH regulates multiple metabolic activities within cells linked to the metabolism and breakdown of macromolecules, such as carbohydrates, proteins, lipids, and damaged cellular organelles, to maintain homeostasis in various conditions [64,65]. TH plays an important role in hepatic lipid metabolism [66]. According to published reports, individuals with obesity are more likely to have hypothyroidism than those with a normal BMI [67,68,69]. These findings strengthen the hypothesis that TH therapy might be of therapeutic benefit in patients with MASLD with or without hypothyroidism [56].
2. Mechanisms of Action of TH
The thyroid hormone receptor (TR), a nuclear receptor, controls T3 activity by acting as a T3-inducible transcription factor. TR has two main isoforms: TRα and TRβ, and its expression varies by tissue. TRα receptor is commonly found in the heart, brain, and bone, whereas TRβ is predominantly found in the liver and kidney. TR interacts with thyroid hormone response elements (TREs) in target gene regulatory domains as a heterodimer with another nuclear receptor, the retinoid X receptor (RXR). TR’s recruitment of coregulator proteins regulates target gene expression. The TR/RXR heterodimer binds the nuclear receptor corepressor and silencing mediator of retinoid and TR to inhibit gene transcription via histone deacetylation in the absence of T3. Coactivators are enrolled when T3 is present, whereas corepressors are dismissed, and TH-responsive gene expression occurs [70]. TH promotes de novo lipogenesis in the liver by recruiting the transcription factor ChREBP to the promoters of lipogenic genes via the T3 receptor TRβ1. The lipogenic genes involved are Acetyl-CoA carboxylase alpha (ACACA), fatty acid synthase (FASN), and thyroid hormone-responsive (THRSP).
3. TH and Its Isoform
THs are synthesized and secreted by the thyroid gland and are required for the control of numerous metabolic processes. The thyroid gland utilizes thyroid follicles as fundamental structures to concentrate iodide and produce the primary THs, namely, 3,3′,5,5′-tetraiodo-L-thyronine (T4) and 3,5,3′-triiodo-L-thyronine (T3) [71]. The anterior pituitary’s thyrotrophs, which release thyroid-stimulating hormone (TSH), regulate TH (mainly T4) secretion from the thyroid gland. By synthesizing and releasing iodinated THs into the bloodstream, the thyroid controls numerous physiological processes in the liver, adipose tissue, central nervous system, cardiovascular system, and musculoskeletal system [72]. Iodothyronine deiodinases (DIO1, DIO2, and DIO3) in extrathyroidal tissue regulate T4 to T3 conversion. Both DIO1 and DIO2 convert circulating T4 to the bioactive TH form, i.e., T3. DIO3 reduces intracellular thyroid by converting T4 and T3 to reverse T3 (rT3) and T2 [73]. Recent studies suggest that T2 has tissue-specific TH activity [18,74].
T3, the most active form of TH, binds to two nuclear hormone receptor isoforms (TRα and TRβ). These receptors function as ligand-inducible transcription factors that interact with TH response elements (TREs) found in target gene promoters, enhancers, and intronic regions [64,75]. TRα is the predominant isoform found in the heart, brain, and bone, whereas TRβ is present dominantly in the liver, accounting for more than 90% of TRs in this tissue [65]. TR subtypes are encoded by two genes. The first gene, TRα, is linked to the c-erbA gene on chromosome 17 [76]. The transcription of C-erbA produces three mRNAs: one full-length TRα1, and the other two variants that code for proteins which do not bind with TH [77,78]. The second gene, THRβ, is located on chromosome 3 and shares DNA with c-erbA. TRα1 mRNA contains numerous alternative start sites, resulting in the translation of three shorter isoforms (p43, p30, and p28) based on their kilo Dalton sizes (Figure 1) [76,79,80]. The mitochondrion is a primary site of TH accumulation within the cell [81,82,83]. TR1α p43 is directed to the mitochondrial matrix, whereas TR1α p28 is particularly directed to the mitochondrial inner membrane [81,84,85]. Over the past decacde, it has become clear that T3 not only exerts its effects through a genomic mechanism but also through nongenomic mechanisms mediated by the mitochondrial thyroid receptor isoforms [86]. The nongenomic effects of TH on mitochondrial FAO are the least studied (and least understood) among the metabolic effects of thyroid hormones. In a collaborative study, researchers conducted in vitro studies and discovered that the T3-induced increase in mitochondrial FAO is, at least in part, mediated by increased levels of MTP due to increased stability and decreased turnover of the MTP complex [87]. Data also suggest that the mitochondrial shortened thyroid receptor isoform p43 (mTR) in particular plays an important role in mediating the T3-induced increase in MTP stability [87].
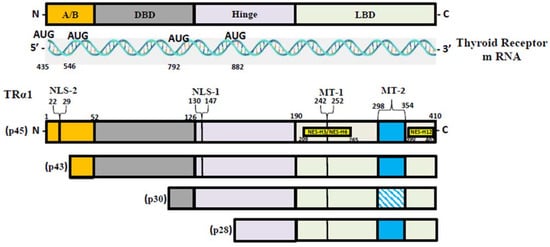
Figure 1. TR-α isoforms. Nuclear localization signals (NLS), nuclear export signals (NES), and mitochondrial targeting signals (MT) are shown in TRα1, TRβ1, and TRβ2. Localization signals are positioned in reference to the individual TR domains: N-terminal A/B domain (A/B); DNA-binding domain (DBE); Ligand-binding domain (LBD).
4. TH and MASLD
TH has been linked to the pathogenesis of MASLD; reduced TH levels have been frequently reported in MASLD patients [88]. According to animal studies, moderate hypothyroidism has been shown to be associated with a higher risk of MASLD [89]. Translational studies of liver transcriptomes from individuals with MASLD undergoing bariatric surgery revealed a decrease in the expression of genes involved in RNA metabolism, protein catabolism, and energy metabolism. These genes, which are controlled by THs under normal physiological conditions, have been shown to have lower expression levels in individuals with MASLD [90]. In rats and humans, lower intrahepatic TH levels in MASLD has been reported [88,91]. TH not only increases de novo lipogenesis (DNL) and improves hepatic insulin sensitivity; it also decreases hepatic gluconeogenesis in hepatocytes. Furthermore, TH promotes lipid export and oxidation [92,93]. The regulation of lipid and glucose metabolism is carried out by the TH receptors, which have direct and indirect effects by interacting with other nuclear receptors such as the peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR), and bile acid signaling pathways [92]. Several observational studies reported a link between increased serum TSH levels and the presence and severity of MASLD [23,94]. In recently published studies, the regulation of hepatic autophagy and mitochondrial metabolism by TH have been described as crucial steps in hepatic triglyceride metabolism [95,96].
4.1. TH and FAO
As shown in Figure 2, thyroid hormone increases FAO oxidation, TCA cycle, and oxidative phosphorylation (OXPHOS) via genomic and nongenomic mechanisms. Thyroid hormone increases expression of PGC1 alpha [97,98], which is a master regulator of mitochondrial function. In addition, there is evidence that TH isoform 43 interacts with MTP and improves its activity by improving its stability [87]. TH has been shown to increase fatty acid import, FAO, and oxygen uptake in isolated mitochondria [99]. Previous work suggests that T2 treatment increased FAO when palmitoyl-CoA was utilized as a substrate rather than palmitoyl-carnitine, suggesting that carnitine-palmitoyltransferase 1 (CPT1) could be a potential T2 target, which was further verified by assessing CPT activity [100]. In isolated mitochondria, TH treatment increased the activity of mitochondrial thioesterase, an enzyme responsible for converting acyl-CoA to fatty acid and CoA [86]. Researchers have recently shown that low-dose T3 treatment in mice increased FAO in both chow- and western-diet-fed-animals [101]. The ETC and the tricarboxylic acid (TCA) cycle also play key roles in FAO [96,102], and CPT1 levels are increased indirectly by TH via surtuin 1 (SIRT1) and PPARα [92,103]. TH further increases the amounts of other mitochondrial enzymes required for FAO such as medium-chain acyl-CoA dehydrogenase (MCAD), pyruvate dehydrogenase kinase, and mitochondrial uncoupling protein 2 (UCP2) [104,105,106]. T2 treatment has been shown to promote hepatic FAO in liver mitochondria, increase downstream respiratory activity, increase proton leak, and reduce oxidative stress in the liver mitochondria without causing thyrotoxicity [107,108].
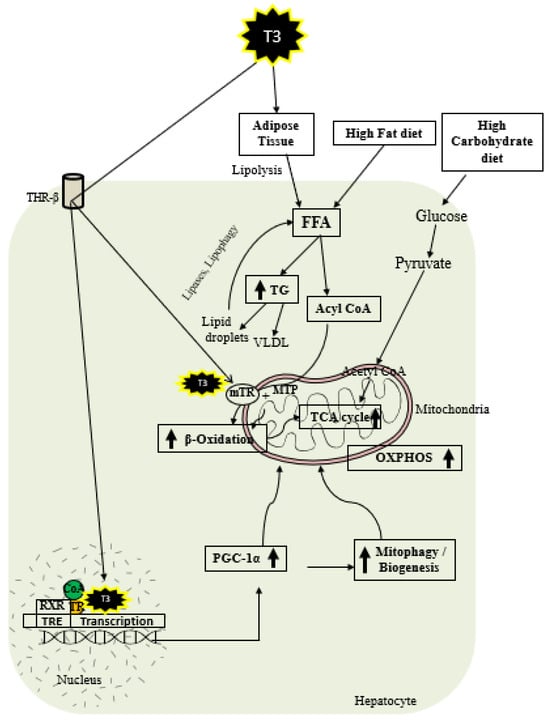
Figure 2. Schematic representations of TH metabolic effects in hepatocytes.
4.2. TH and Mitochondrial Biogenesis
TH exerts multiple actions at a molecular level aimed at increasing the number of mitochondria. As shown in Figure 2, T3 promotes mitophagy and mitochondrial biogenesis via peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) [48,109]. TH stimulates mitochondrial biogenesis by inducing PGC1α gene expression, which stimulates the transcription of nuclear respiratory factor 1 (NRF1) and mitochondrial transcription factor A (mtTFA) [21]. SIRT1 is activated by TH, which deacetylates PGC1α and increases its ability to bind the regulatory areas of mitochondrial synthesis and function genes [103]. Further, T3 also increases the expression and activation of Unc-51-like autophagy activating kinase 1 (ULK1), which improves the dynamin-related protein 1 (DRP1)-mediated mitochondrial fission, activation, and association of FUN14 domain-containing 1 (FUNDC1) with LC3B, and p62 translocation to mitochondria in hepatic cells [109,110]. T3-mediated mitophagy activation is required for mitochondrial oxidative phosphorylation system (OXPHOS) stimulation [109]. Additionally, T3 promotes mitochondrial biogenesis [110] and increases the rates of mitophagy and mitochondrial synthesis, both of which contribute to enhanced mitochondrial activity and fatty acid β-oxidation.
4.3. TH and Mitophagy
To minimize cellular harm caused by reactive oxygen species (ROS), TH induces protective autophagy of mitochondria, a process called mitophagy [109]. Mitophagy is initiated by excessive ROS production from mitochondria, leading to the release of intracellular Ca2+, activation of calcium/calmodulin-dependent protein kinase 2 (CAMMK2), phosphorylation of AMP-activated protein kinase (AMPK), and subsequent activation and translocation of ULK1 to the mitochondria after AMPK phosphorylation [109]. Elevated ROS levels are perpetuators of mitophagy, which ensures the preservation of mitochondrial quality required for β-oxidation of fatty acids and oxidative phosphorylation. Mitophagy induced by ROS generation selectively sequesters damaged mitochondria to be removed from the cell, preventing additional oxidative damage and cell death [109]. Recent research suggests that PGC-1α is involved in the complex regulation of mitochondrial quality beyond biogenesis, including mitochondrial network dynamics and autophagic removal of damaged mitochondria [111].
5. Potential Therapeutic Use of TH and Its Analogs in MASLD
In recent years, THs, as well as the TR-β agonists and additional liver-specific analogs, have been studied as a potential MASLD treatment [23]. Research from previous studies reported the use of T3 to promote weight loss in patients with obesity and to treat hypercholesterolemia [112]. T3 injections given daily intraperitoneally (ip) to ob/ob mice have been found to decrease both body weight and fat while increasing oxygen intake and oxidative metabolism [113]. GC-1, a novel TR-β agonist, has been reported to reduce the development of hepatic steatosis and lipid peroxidation in mice [46] and decrease hepatic TG levels with no major side effects [114,115]. MB07811, another orally administered TR-β agonist, has been shown to prevent hepatic steatosis in rats and mice via boosting β-oxidation and mitochondrial respiration rates, lowering hepatic TG levels and stimulating CPT1α expression [47]. Resmetirom (MGL-3196) has been shown to be effective in lowering hepatic TG, lipid peroxidation, ALT, steatosis, inflammation, and fibrosis in animal models [116,117]. Increased mitochondrial β-oxidation has been suggested to be one of the mechanisms by which Resmetirom decreases liver fat [118]. VK2809 therapy has been shown to reduce hepatic lipid accumulation in a glycogen storage disease Ia (GSD1a) mouse model by restoring the autophagy, mitochondrial biogenesis, and β-oxidation of fatty acids [119]. KB2115, a TR-β agonist, has been reported to decrease the total and low-density lipoprotein (LDL) cholesterol levels in the blood and to prevent the development of hepatic steatosis [120]. Ongoing research has demonstrated that low-dose T3 is effective in increasing hepatic mitochondrial FAO and reversing MASLD in mice [121]. Based on these results, researchers recently initiated a randomized double-blinded placebo-controlled clinical trial to test whether low-dose thyroxine (T4) is effective in improving the histological features in Veterans with biopsy proven MASH (National Library of Medicine NCT05526144) [122]. Taken all together, the results of the above studies demonstrate that TH therapy might be an effective strategy in treating MASLD/MASH. The use of TH and its analogs in preclinical and clinical research is summarized in Table 1.
Table 1. Effects of TH and its analogues in MASLD.
| Compound | Model and Dose | Study Findings | MASLD Impact | References |
|---|---|---|---|---|
| Animal Studies | ||||
| TH | ||||
| T2 | Hepatocyte isolated from Wistar rats; 10−7 to 10−5 M | Reduction of acyl-CoA oxidase and peroxisomal β-oxidation | Reduction of hepatic lipid accumulation | [123] |
| T2 | C57BL/6J mice; 2.5 µg/100 g; ip | Increased fatty acid oxidation and decreased lipogenesis | Inhibition of fat accumulation in liver | [124] |
| T2 | Male wistar rats; 25 µg/100 g; ip |
Reduced hepatic fatty accumulation, enhanced fatty acid oxidation rate and carnitine palmitoyl transferase activity | Activates mitochondrial processes, reverses hepatic steatosis | [125] |
| T2 | Rats injected with 25 µg/100 g; ip |
Reduction in Serum TG and cholesterol | Prevents fatty liver by increasing fatty oxidation | [107] |
| T3 | ob/ob mice; 25 µg/100 g; ip | Lowered body weight and fat, increased oxidative metabolism | Increased oxidative metabolism in brown adipose tissue and liver | [113] |
| T3 | Male wistar rats; 25 µg/100 g; ip | Promotes fatty acid peroxisomal and mitochondrial β-oxidation | Prevents hepatic fat accumulation by increasing β-oxidation | [46] |
| T2 and T3 | Wistar rats; 25 and 2.5 µg/100 g; ip | Increased CPT-1 levels | Lowering hepatic lipid content, induced autophagy and intra-hepatic acylcarnitine flux | [126] |
| T4 | Male C57BI/6J mice; | Decreased hepatic triglyceride and cholesterol | Reduce hepatosteatosis and prevent MASH progression | [127] |
| Thyroid hormome analogues | ||||
| T3 and TRβ agonist GC-1 | Male fischer rats; 4 and 5 mg/kg; ip |
Marked fatty liver with mild hepatitis | Prevents fat accumulation by increasing mitochondrial and peroxisomal oxidation, complete regression of liver steatosis | [46] |
| TRβ agonist GC-1 | Male sprague Dawley rats; 1 µg/kg; oral gavage | Reduction in hepatic TG levels | Treatment of obesity and hypercholesterolemia | [114] |
| MB07811 | Male sprague Dawley rats, ob/ob mice; 1 to 50 mg/kg; oral gavage | Prevents hepatic steatosis, reduced plasma FFA and triglycerides | Increased hepatic fatty acid β-oxidation and mitochondrial respiration rates, as well as lower hepatic triglyceride levels and stimulation of CPT1α expression | [47] |
| Resmetirom (MGL-3196) | C57BI/6J mice; 3 mg/kg for 8 weeks by oral gavage |
Lower hepatic triglycerides, lipid peroxidation, steatosis, inflammation and fibrosis |
Improvement in systemic and hepatic metabolism | [116] |
| VK2809 | GSDIa mouse model; 10 mg/kg; Subcutaneously | Restoring autophagy, mitochondrial biogenesis, and β-oxidation of fatty acids | Reduced hepatic lipid accumulation | [119] |
| GC-1 and KB-2115 | Male Sprague-Dawley rats; 164 and 100 µg/kg; ip | Increased white adipose tissue lipolysis | Reduced hepatic steatosis | [128] |
| TG68 | C57BL mice; 2.8 mg/kg in drinking water | Reduction in liver weight, hepatic steatosis and triglycerides | Can be used in MASLD | [129] |
| TRC150094 | Male wistar rats for 8 weeks; ip injection (0.750 mg/100 g b wgt | Reduction of Fat accumulation | Can be used in MASLD | [130] |
| Clinical Trials | ||||
| TH | ||||
| MGL-3196 (Resmetirom) | 36 weeks randomized trial in patients with biopsy proven MASH with fibrosis given 80 mg orally daily | Significant reduction of hepatic fat, liver enzymes, lipoprotein, inflammation and fibrosis. | Patients showed reduction of hepatic fat compared to placebo, adverse events were mild and moderate | [131] |
| 2 weeks randomized trial with 0.25 to 200 mg/day | Significant reduction of total cholesterol and triglycerides |
Safe and showed beneficial effect on lipid parameters | [132] | |
| KB2115 (eprotirome) |
5-day randomized trial in patients given 50 to 2000 µg orally daily |
Reduction in serum TC and LDL in overweight patients |
Reduced body weight | [120] |
| VK2809 | 12-week study of low dose of 5 mg in patients |
Reduction in LDL levels | Improvements in liver fat content in patients with MASLD | [18] |
| Levothyroxine (T4) | Patients with type 2 diabetes and steatosis given 18.75 µg/day | Low dose T4 decreased lipid content in euthyroid male patients with type 2 diabetes mellitus. | Safety and efficacy of TH therapy for MASLD in men | [91] |
| DITPA | 8-week randomized trial in patients with dose from 90 till 360 mg/d | Lowered serum cholesterol and decrease in triglycerides |
Reduced body weight | [133] |
This entry is adapted from the peer-reviewed paper 10.3390/cells12242806
This entry is offline, you can click here to edit this entry!