Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pediatrics
Wilms tumor (or nephroblastoma) is a malignant and solid neoplasm that derives from the primitive renal bud. It represents the most frequent primary tumor of the urogenital tract in childhood, and treatment consists of surgery and chemo-radiotherapy.
- Wilms
- nephroblastoma
- pediatric urogenital cancer
- cancer
1. General and Epidemiological Profiles
Wilms tumor (or nephroblastoma) is a solid malignant neoplasm that derives from the primitive renal bud. Wilms tumor represents the most frequent primary renal form of the urogenital tract in childhood [1] and can be unilateral (in 90–95%), bilateral or multicentric (in forms related to genetic factors), with both synchronous and metachronous presentation [2]. The prevalence of this tumor ranges between 2% and 6% among all childhood neoplasms [3], with an estimated worldwide prevalence of just over 1:10,000 children in Europe and North America and 4:10,000 in Asian countries [4,5], based on three characteristics that influence its epidemiological trend: (a) age (this tumor mainly affects children under the age of 15 with an average age at diagnosis between 2 and 5 years, and in general as many as 75% of cases occur before the age of 5); (b) gender (it is more frequent among females than males); (c) country of origin (it has a higher incidence in individuals of African origin, while it has a much lower incidence among Asians, with Europeans being in an intermediate position [6,7,8].
2. Pathology
From a macroscopic point of view, the tumor appears as a mass with well-defined margins, single or, more rarely, multiple, of a soft consistency; when cut, it appears grayish in color and homogeneous in appearance, although cysts, areas of necrosis or bleeding may be found inside. On the other hand, microscopic examination can reveal various aspects which recall the various stages of the embryological development of the kidney. (Figure 1) The most common picture consists of three cell types (epithelial, stromal and blastema) in various percentages. Each of these cell types can be present in different stages of differentiation: for example, epithelial cells can differentiate into tubular or glomerular cells, while stromal cells can remain undifferentiated or take on a fibrotic, myxoid or even skeletal muscle cell appearance (Figure 2) [9].
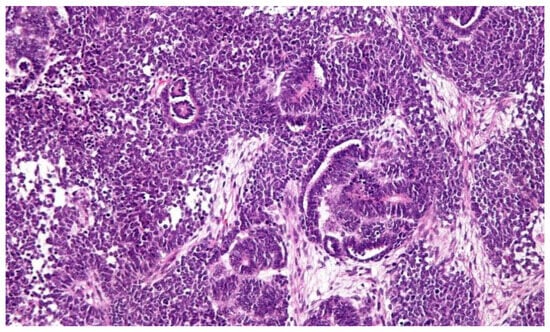
Figure 1. Classical histological features of WT include a triphasic pattern of epithelial, stromal and blastemal components. The image and description are from the AFIP Atlas of Tumor Pathology, according to entry #407018 in Pathology Education Instructional Resource. The Armed Forces Institute of Pathology Electronic Fascicles (CD-ROM Version of the Atlas of Tumor Pathology) contains U.S. Government work which may be used without restriction.
Figure 2. Wilms tumor. Scanning electron micrograph of cell culture about Wilms tumor, which is a human tumor that, like typical tumors, when attached to a surface is round. After it has been attached, it spreads out. The magnification is ×130. This image is in the public domain and can be freely reused. Source: https://visualsonline.cancer.gov/details.cfm?imageid=1763 (accessed on 7 September 2023).
3. Clinical Profiles
Unlike renal cell carcinoma, which is often asymptomatic, Wilms tumor often manifests itself in the form of a palpable abdominal mass, with a smooth surface and the presence of abdominal pain. Systemic symptoms consist of fever, weight loss and fatigue, and less frequently also hematuria (due to internal bleeding) and hypertension (as a result of renin production by tumor cells or the activation of the renin–angiotensin–aldosterone axis due to reduced blood supply to the kidney when the tumor compresses the renal artery) can be present. Five stages of the disease are generally identified and defined based on the extent of the tumor and its eventual distant spread: [5,9,10,11].
Stage I: (a) The tumor is limited to the kidney. (b) The tumor is present in the perirenal fat but is surrounded by a fibrous (pseudo)capsule. The (pseudo)capsule might be infiltrated by a viable tumor which does not reach the outer surface. (c) The tumor might show protruding (botryoid) growth into the renal pelvis or the ureter but does not infiltrate their walls. (d) The vessels or the soft tissues of the renal sinus are not involved with the tumor. Intrarenal vessel involvement might be present.
Stage II: (a) A viable tumor is present in the perirenal fat and is not covered by a (pseudo)capsule, but it is completely resected (resection margins are clear). (b) The viable tumor infiltrates the soft tissues of the renal sinus. (c) The viable tumor infiltrates the blood and/or lymphatic vessels of the renal sinus or the perirenal tissue, but it is completely resected. (d) The viable tumor infiltrates the wall of the renal pelvis or the ureter. (e) The viable tumor infiltrates the vena cava or adjacent organs (except the adrenal gland) but is completely resected.
Stage III: (a) The viable tumor is present at a resection margin. A nonviable tumor or chemotherapy-induced changes present at a resection margin are not regarded as stage III. (b) Abdominal lymph node involvement is present by either viable or nonviable tumors. (c) Preoperative or intraoperative tumor rupture, if confirmed via microscopic examination (viable tumor at the surface of the specimen at the area of the rupture). (d) The viable or nonviable tumor thrombus is present at the resection margins of the ureter, renal vein or vena cava inferior (always discuss resection margins with the surgeon). (e) The viable or nonviable tumor thrombus, which is attached to the inferior vena cava wall, is removed piecemeal by a surgeon. (f) Wedge or open tumor biopsy before preoperative chemotherapy or surgery. (g) Tumor implants (viable or nonviable) are found anywhere in the abdomen. (h) The tumor (viable or nonviable) penetrates through the peritoneal surface.
Stage IV: Hematogenous metastases (for example, lung, liver, bone and brain) or lymph node metastases outside the abdominopelvic region.
Stage V: Bilateral renal tumors at diagnosis. Each side should be substage according to the above criteria.
Wilms tumor is often associated with other malformations or congenital defects of the urogenital system or other systems. It presents in Beckwith–Wiedemann syndrome, a genetic pathology usually caused by an imprinting defect (defect in the modulation of the expression of a part of the genetic material) or uniparental paternal disomy (when an individual receives two copies of a chromosome (or a whole chromosome and part of the second chromosome) from either parent and no chromosome copy from the other parent) for the 11p15 region, with an incidence of 1:15,000 births [12], or WAGR syndrome, which is characterized by mental retardation, aniridia and genital anomalies [13], or Denys–Drash syndrome, in which male pseudohermaphroditism is observed due to a defect in the development of the gonads and renal insufficiency [14].
4. Therapy
Treatments are modulated according to clinical and anatomopathological variables, as well as according to national protocols specific to each national health system. Generally, the initial treatment of unilateral Wilms tumor is primary surgical resection (with an approach that can be either anticipated or delayed based precisely on each patient’s histologic and clinical outcomes) followed by adjuvant chemotherapy. A select group of younger patients with small tumors can be cured with surgery alone. The type of chemotherapy drug and the duration of therapy depend on the tumor histology and stage. Chemotherapy regimens depend on the risk group but usually consist of actinomycin D (dactinomycin) and vincristine, with or without doxorubicin, or adriamycin. For more aggressive tumors, intensive multi-agent chemotherapy regimens are used. Children with very large unresectable tumors or bilateral tumors are candidates for chemotherapy followed by re-evaluation and resection at a later time. Radiotherapy is given to children who have the disease at a higher stage (stage III and in the presence of distant metastases, usually of the lungs, which do not regress readily with chemotherapy). In most cases, radical nephrectomy is practiced, i.e., the surgical removal of the affected kidney, associated with the resection of the regional and para-aortic lymph nodes ipsilateral to the neoplasm; in bilateral tumors or patients with specific syndromes predisposed to the onset of nephroblastoma, partial nephrectomy is to be preferred whenever feasible [39,40,41,42,43,44]; when possible, especially in the case of bilaterality, preference should be given to even partial preservation of the renal structure, unless clinical conditions permit and the balance with the renal function to be preserved is compatible with possible tumor recurrence [45]. An improvement in the clinical picture and renal parenchyma over historical outcomes emerges in children with the bilateral form (but not in diffuse anaplasia and inhomogeneous tumors) if the treatment approach includes standardized three-drug preoperative chemotherapy, surgical resection within 12 weeks of diagnosis and response and postoperative therapy based on the histologic picture [46,47,48,49,50,51]; still, in the experimental phase, one study showed that concomitant administration of WT1-immunotherapy and standard neoadjuvant therapy (used in breast cancer) was well tolerated and induced WT1-specific antibodies in patients receiving aromatase inhibitors in the neoadjuvant phase (precisely for patients with Wilms tumor); however, in patients receiving neoadjuvant chemotherapy or the trastuzumab-chemotherapy combination, the humoral response was impaired or attenuated, probably due to the co-administration of corticosteroids and/or the chemotherapeutics themselves [52]. One study finally showed that radiofrequency with cryoablation was also effective for this tumor type [53]. To this day, however, immunotherapy and cryotherapy are not yet generally approved therapies in scientific communities such as the Children’s Oncology Group and the Renal Tumor Study Group of the International Society of Pediatric Oncology (SIOP-RTSG), due to the few studies still in the literature, but it is hoped that in the future they may be considered as alternative and/or concomitant therapies to improve the quality of Wilms tumor treatments. [54] Another study has shown that the International Society of Pediatric Oncology (SIOP) protocol for Wilms tumor (WT), in the part that calls for preoperative chemotherapy as an initial approach, does not take into account the risk of histologic misdiagnosis and thus ineffective or even harmful treatment; therefore, it has been shown that fine-needle aspiration cytology (FNAC) can be a useful and feasible technique in children and can confirm the diagnostic suspicion of unilateral WT, avoiding inadequate preoperative chemotherapy in non-Wilms renal cancer. [55] Surgery, chemotherapy and radiotherapy essentially constitute the current treatment modality for Wilms tumor. The guidelines of the National Wilms Tumor Study Group (NWTSG/COG) and the International Society of Pediatric Oncology (SIOP) provide two different strategies for initial treatment, with some differences: in particular, in North America, the NWTSG/guidelines are adopted by the COG, while in Europe, it is usual to use the SIOP guidelines. Despite differences, determined from the individual case, the overall survival rate for patients treated using the two guidelines is similar, i.e., above 90% (in uncomplicated cases or with metastasis) [5]. Differences in approach between the different European and American currents, however, are resolved through the integration of practical experience, keeping in mind the specific characteristics of the patient’s clinical history and the relevant guidelines. In the table, see the different treatments (Table 2):
Table 2. Differences between the COG and SIOP treatment protocols for WT. COG = National Wilms Tumor Study Group (NWTSG)/Children’s Oncology Group; SIOP (The International Society of Pediatric Oncology) [5].
| Type | COG | SIOP |
|---|---|---|
| Surgery | Primary surgery before chemotherapy is recommended. For resectable tumors, preoperative or intraoperative biopsy is not performed, whereas in radical nephrectomy and lymph nodes, harvesting is performed through a transabdominal incision. To prevent tumor leakage, en bloc resection can be performed. The resection of a primary renal tumor should be considered even if at a stage IV disease (with metastases); renal-sparing surgery is not recommended, except in children with a solitary kidney, a predisposition to bilateral tumors or a horseshoe kidney, or in infants with Denys–Drash or Frasier syndrome (in order to delay the need for dialysis). | Radical nephrectomy of the tumor, performed after preoperative chemotherapy, is recommended. Lymph node sampling is important for staging, and the sampling of seven locoregional lymph nodes is necessary for accurate staging. Nephron-sparing surgery is used for nonsyndromic unilateral Wilms tumors, provided the following clinical conditions are met: (a) small tumor volume (<300 mL); (b) the expectation of substantial residual renal function in patients who have never had lymph node involvement. |
| Chemotherapy | Surgery is recommended as initial therapy before chemotherapy. Preoperative chemotherapy is indicated only in the following conditions: (a) with an inoperable Wilms tumor type; (b) with a solitary kidney; (c) with bilateral synchronous Wilms tumor; (d) a tumor thrombus in the inferior vena cava, extending above the level of the hepatic veins; (e) a tumor involving contiguous structures, whereby the removal of the kidney requires the removal of other organs (such as the spleen, pancreas or colon; (f) stage IV (with extensive pulmonary metastases). | Preoperative chemotherapy is recommended for all patients after diagnosis. For patients with unilateral localized tumors, a 4-week pretreatment is administered using vincristine (weekly) and dactinomycin (biweekly), while for patients with bilateral tumors, vincristine–dactinomycin is recommended for no more than 9 to 12 weeks (in some patients, doxorubicin is added as a reinforcer). Again, for patients with metastases, a regimen including 6 weeks of vincristine–#dactinomycin (as described above) and doxorubicin at weeks 1 and 5 is given. |
| Postoperative chemotherapy | It is recommended that postoperative chemo-therapy be used routinely in all patients with Wilms tumor except those at very low risk (those less than 2 years of age at diagnosis with a tumor, with favorable stage I histology, weighing less than 550 g, with sampling and with confirmed negative lymph nodes). | Postoperative chemotherapy is recommended in all patients with Wilms tumor except those with low-risk stage I tumors. |
| Postoperative radiation | Postoperative irradiation in the tumor bed is recommended for all patients with stage III cancer. | Radiation therapy of the whole abdomen is recommended for patients with intermediate-histology or high-risk tumors and with major tumor rupture preoperatively or intraoperatively, or with macroscopic peritoneal deposits. Lung radiotherapy is indicated for lung metastases without complete response until the 10th postoperative week. Patients with a complete response after induction chemotherapy with or without surgery do not need pulmonary radiotherapy. Patients with viable metastases at surgery or with high-risk histology require pulmonary radiation therapy. Whole-lung irradiation is recommended for patients who did not receive lung irradiation during first-line treatment, regardless of histology. |
| Recurrent WT | Wilms tumors with characteristic high recurrence are divided into three risk groups: (1) standard risk; (2) high risk; (3) very high risk. In the first case (1), surgery (when possible), radiotherapy and chemotherapy (alternating cycles of vincristine/do-xorubicin/cyclophosphamide and etoposide/cyclophosphamide) are used. In the second and third cases (2,3), chemotherapy (alternating cycles of cyclophosphamide/ethoposide and carboplatin/ethoposide), surgery and/or radiotherapy and hematopoietic stem cell transplantation are recommended. | Patients with Wilms tumor are classified into AA, BB and CC, but essentially nothing changes from the previous classification. For the former (AA), only vincristine and/or dactinomycin is used as first-line treatment (without radiotherapy), with a four-drug regimen (combinations of doxorubicin and/or cyclophosphamide and carboplatin and/or etoposide); for the second group (BB), an intensive reinduction regimen (including the combination of etoposide and carboplatin with phosphamide or cyclophosphamide) is administered, followed by high-dose melphalan and autologous stem cell rescue or two more rounds of reinduction; for the third group (CC), camptothecins (irinotecan or topotecan) or new biologic compounds are recommended. |
| Stage V—WT | Both the COG and SIOP recommend preoperative chemotherapy and resection for bilateral WT. Bilateral renal-sparing surgery can be performed in patients with synchronous bilateral WT. Renal parenchyma sparing may help preserve renal function in these children. Renal transplantation is recommended and is usually delayed for 1–2 years without evidence of relapse. The SIOP also suggests that preoperative chemotherapy should be limited to no longer than 12 weeks, with time intervals for evaluation fixed to 6 weeks. | |
| Accepted Chemotherapy Regimens for Wilms Tumor |
|
|
The current research group, which replaces the National Wilms Tumor Study Group (NWRSG), is the Children’s Oncology Group Renal Tumor Committee (COGRTC), adhering to the principles enucleated in the previous description [60].
The criteria for subclassifying pretreated WTs are as follows: The completely necrotic type shows no viable tumor elements. If more than 66% (two-thirds) of the tumor is non-viable (i.e., shows chemotherapy-induced changes), it is regarded as a regressive type, irrespective of the presence of remaining viable tumor components. If a viable tumor comprises more than one-third of the tumor mass, subtyping depends on the percentage of viable components: in mixed type, none of the components comprise more than 66% of the tumor; in epithelial (or stromal) type, in addition to having more than 66% of the tumor being composed of epithelial (or stromal) elements, the finding of only up to 10% of blastema is allowed (if the finding is more, then the tumor is subclassified as mixed type). Finally, preoperative chemotherapy is recommended for children with Wilms tumor with intravascular extension, as prolonged chemotherapy may improve resectability but increases tumor adhesion to the vascular endothelium, precluding complete resection. To evaluate the optimal duration of preoperative treatment, a study focused on overall survival (OS) and event-free survival (EFS), tumor regression, complete resection and cavectomy demonstrated that there is no evidence that cycle-prolonged periods of neoadjuvant chemotherapy confer additional benefits in treatment and are therefore not recommended [61].
The classic triphasic Wilms tumor does not present diagnostic difficulties for pathologists, but when only one component is present (especially if small), the differential diagnosis may include renal cell carcinoma, metanephric adenoma and hyperplastic nephrogenic remnant, sarcoma of the kidney, mesoblastic nephroma and synovial sarcoma. Even pure Wilms tumor of the blastemal type may be difficult to distinguish from other embryonic “small round blue cell tumors”, including neuroblastoma, primitive neuroectodermal tumor/Ewing’s sarcoma, desmoplastic small round cell tumor and lymphoma. There are several reasons why making the correct diagnosis of Wilms tumor can be difficult: (1) the rarity of pediatric renal tumors results in a lack of experience with these entities for most pathologists; (2) the presence of different Wilms subtypes (morphological heterogeneity); (3) the morphological aspects can vary considerably from case to case; (4) the histological patterns of some Wilms subtypes may initially appear similar to those of other rare pediatric renal tumors; (5) the lack of clear-cut differential criteria distinguishing Wilms from nephrogenic remnants (NR), especially in limited biopsy material; (6) tumor evaluation and local pathological stage determination are multistep and time-consuming processes [62].
This entry is adapted from the peer-reviewed paper 10.3390/surgeries4040064
This entry is offline, you can click here to edit this entry!