1. Retinal Degeneration in Alzheimer’s Disease (AD)
Numerous studies have highlighted visual impairments in individuals with AD, often manifesting earlier than other dementia-related symptoms [
1,
2,
3]. Ashok et al. [
4] elucidated that these visual changes arise not solely from pathological developments in the visual cortex but also from the loss of Retinal Ganglion Cells (RGC) and degeneration associated with age-related macular degeneration (AMD) [
5,
6]. Intermediate hard drusen were commonly found in the temporal region of AD eyes compared to older normal eyes. A recent study has shown that, starting from postmortem brain and eye tissues samples from AD donors, there was a significant relationship between cerebral amyloid angiopathy (CAA) level and number of temporal intermediate hard drusen [
7]. Other retinal manifestations in AD encompass diminished macular blood flow and astrogliosis [
8]. Interestingly, certain eye diseases exhibit hallmark AD histopathological changes, indicating some overlap. For instance, amyloid-beta (Aβ) and iron accumulate in drusen [
9,
10,
11], a defining sign of AMD, while phosphorylated tau (p-tau) and Aβ are found in RGCs, which are primarily affected in glaucoma.
However, in sporadic AD (SAD), which accounts for 85% of AD cases [
12], the accumulation of Aβ and p-tau in RGCs is not consistently observed [
13,
14,
15]. Vision-related abnormalities are prevalent in AD [
16]. A decrease in the retinal nerve fiber layer (RNFL) thickness, especially in the inferior and superior quadrants [
17,
18,
19,
20], due to selective RGC loss, is now recognized as a potential AD diagnostic marker [
21,
22].
This marker can be non-invasively detected using optical coherence tomography (OCT) and electroretinography (ERG). However, some authors argue that these tests lack the specificity and sensitivity needed for broad clinical applications [
19,
23,
24,
25].
The processing of amyloid precursor protein (APP) in the eye, and its impairment in the brain remains a topic of debate, as does the efficiency of Aβ clearance mechanisms in both the brain and the eye. A significant correlation has been observed between Aβ and degeneration in the RGC layer, photoreceptors, and the retinal pigmented epithelium (RPE) [
4,
26]. Conversely, p-Tau has been identified from the ganglion cell to the outer plexiform layer [
27,
28]. While the presence of Aβ in drusen is established, the primary factor causing APP metabolism imbalance in RPE cells remains unidentified. Zhao et al. [
29] noted that during normal aging, the production and secretion of Aβ1−42 increase in RPE cells. This leads to its deposition at the interface of RPE cells and the outer segments of photoreceptors and in the subretinal space, where it should be cleared by microglia. However, excessive Aβ expression in RPE cells results in AMD-like pathology [
30], causing microglia to accumulate Aβ and other cellular debris, leading to inflammation and typical AMD drusen deposition. Rong et al.’s meta-analysis [
31] supports the role of Aβ in AMD pathogenesis, highlighting a significant AD–AMD link [
32].
Glaucomatous degeneration of RGCs is linked with p-tau, potentially overexpressed due to increased shear stress and other cytoskeletal proteins via the ROCK kinase pathway. AD also affects other eye regions beyond the pars nervosa. The cornea exhibits increased sensitivity [
33] and decreased thickness [
34,
35]. Pupillary abnormalities in AD patients include slower responses to light and target detection tasks [
36], exaggerated reactions to dilute tropicamide [
37,
38], and a reduced baseline size [
39]. Additionally, during cognitive tasks, the pupil diameter appears enlarged [
40]. Some AD patients develop equatorial supranuclear cataracts [
41,
42], but the link between lens opacity and AD is debated. While Sun et al. [
43] found no optic nerve axonal damage in AD patients, other studies describe axonal degeneration [
44,
45] and lamina cribrosa thinning [
35]. Bayer et al. and Wostyn et al. [
46,
47] reported elevated intraocular pressure in AD patients. Expectedly, these changes lead to various visual dysfunctions, including impaired color vision, contrast sensitivity, visual acuity, and visual integration, along with visuospatial impairments, reduced macular thickness, visual field loss, and visuomotor deficits [
48].
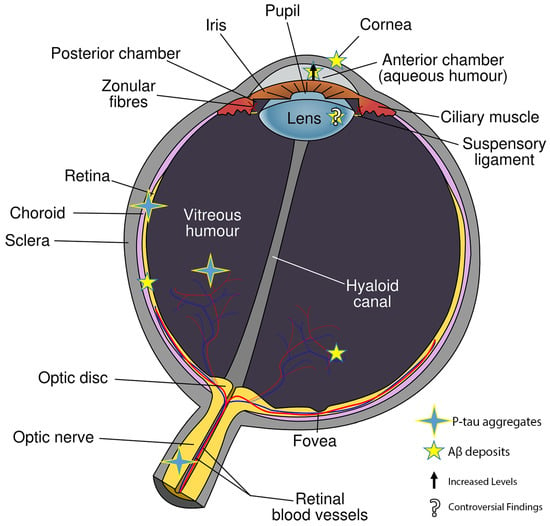
2. Histopathological Alterations in Different Eye Cytotypes Due to β-Amyloid Production
During the progression of AD, multiple eye cell types, both neural and non-neural, exhibit distinct metabolic changes, which subsequently influence gene expression. While these changes have been documented (
Figure 1), a comprehensive understanding remains elusive. It is widely believed that a chronic elevation of intraocular pressure (IOP) may instigate the accumulation of not only p-tau in RGCs, but also Aβ, leading to cell death. The processing of APP appears to be consistent in the retina and other eye cell types. This is further supported by the presence of soluble APPα, APPβ, and pathogenic Aβ in the vitreous humor (VH) and aqueous humor (AH), as well as in cerebrospinal fluid (CSF) [
49]. Aβ deposits span across all retina layers, from the nerve fiber layer to the photoreceptor layer. Analogous to the brain, retinal Aβ fosters tau protein hyperphosphorylation, leading to its aggregation in structures like neurofibrillary tangles (NFT) [
15,
50,
51]. Elevated IOP has been linked to increased tau oligomerization, resulting in RGC death. The suppression of tau using short interfering RNA (shRNA) has been shown to rescue RGCs, further emphasizing the relationship between p-tau accumulation and cell death [
52].
Figure 1. Where Aβ and p-tau were found in eye tissues.
Tau’s role in stabilizing microtubules means that its hyperphosphorylation and aggregation can disrupt anterograde axonal transport, affecting several neuronal functions. For instance, it can hinder mitochondrial transport, leading to energy depletion and the generation of reactive oxygen species (ROS) [
53]. In the cornea, both epithelial cells and fibroblasts seem to be involved. Fibroblasts show increased expression of ADAM-10 (α-secretase, the Amyloid Precursor Protein processing enzyme which starts non-amyloidogenic pathway cleaving at residue 697) and BACE-1 (β-secretase, the Amyloid Precursor Protein processing enzyme which starts amyloidogenic pathway cleaving at residue 11) [
54], while epithelial cells exhibit heightened APP expression, Aβ accumulation, morphological changes, and an increased rate of apoptosis [
34]. Both cell types express APP and produce Aβ [
55].
In the retina, notable histopathological markers include a shift towards anaerobic metabolism [
56,
57], the presence of Aβ plaques [
14,
58], p-tau deposits [
13,
52], vascular changes [
59,
60], and blood–retinal barrier disruptions [
61]. Aβ also accumulates in the retinal microvasculature and pericytes [
61]. Significant gene expression changes have been observed, such as increased levels of retinal vascular β40 and β42 Aβ fragments and decreased levels of vascular PDGFR-β and LDL-1 [
61]. The lens’s involvement in AD is debated; while some researchers have observed increased Aβ aggregation [
42,
62] and presenilin expression [
63], others have not [
54,
64]. The aqueous humor has shown elevated Aβ levels [
65,
66], whereas the vitreous humor is rich in AD-associated proteins [
49,
66]. The choroid appears thinner [
67,
68], and tau deposition has been observed in the optic nerve [
69].
3. Retinal Histopathological Abnormalities in AD Mouse Models
Various AD animal models have exhibited retinal Aβ deposits, often accompanied by apoptotic RGCs and axonal degeneration [
70,
71]. For example, in 3xTG-AD, APP-PS1ΔE9, and APPswe/PS1ΔE9 mouse models, which are known to develop Aβ deposits in the brain, there have been observations of retinal Aβ oligomers and thinning of the retinal nerve fiber layer (RNFL) [
6,
72,
73]. In a glaucoma rat model, the retinal threshold for Aβ1−42 increased in response to elevated intraocular pressure (IOP), aging, and light exposure, leading to RGC apoptosis [
52]. Walsh et al. [
74] replicated this condition by administering intravitreal injections of Aβ1−42. Their research demonstrated that using agents that either reduce toxic Aβ fragments or induce anti-amyloidogenic mutations can protect RGCs. Lastly, in tauopathy mouse models (where mutations are introduced in MAPT, the tau gene), a direct relationship was established between p-tau, Aβ deposits, and RGC death, highlighting the three primary contributors to the retinal pathology associated with AD [
75].
4. The Role of the Prion Protein in Retinal Allostasis
In the progression of Alzheimer-Perusini’s disease, the prion protein (PrP) plays a multifaceted role. As described by H. H. Jarosz-Griffiths et al. [
76], its function can be likened to the “
Ugly” character in Ennio Morricone’s renowned film. PrP can act as a conduit for the cytotoxic effects of Amyloid-beta (Aβ), serving as a scavenger receptor. Conversely, it can also block Aβ-derived fragments, preventing their aggregation in the extracellular space, thus acting as a decoy [
12]. Currently, two Aβ binding sites on the PrP
C structure have been identified: one spanning amino acid residues 95–105 and the other 23–27 [
77]. However, only blocking the larger site (residues 95–105) protects neurons from Aβ’s harmful effects. When Aβ oligomers bind to PrP, the downstream tyrosine kinase Fyn pathway is activated, leading to increasingly severe synaptic alterations until their eventual destruction [
78].
Like APP, PrP
C undergoes various cleavage patterns, termed α-, β-, or γ-cleavage. α-cleavage is facilitated by ADAM17 [
79], which belongs to the α-secretase family (also known as TNFα-cleaving enzyme, TACE). This cleavage results in the release of the N-terminal soluble fragment N1 into the extracellular space, while the C-terminal fragment remains attached as C1 via a GPI anchor. Given that C1 lacks the primary Aβ-binding site, this post-translational modification is believed to protect against Aβ-induced toxicity. Conversely, β-cleavage produces a membrane-anchored C-terminal fragment, C2, which contains the entire primary Aβ-binding region, and a released N-terminal fragment N2, which contains the secondary Aβ-binding site spanning residues 23−27. Thus, C2 fragments may facilitate Aβ neurotoxic transmission. γ-cleavage results in a soluble form of PrP
C. In neurons, the majority (65–80%) of PrP
C cleavage occurs at position 111/112, releasing the N1 fragment (α-cleavage). However, in the retina, β-cleavage predominates, primarily due to oxidative stress. This cleavage pattern is believed to protect cells from oxidative damage. The same trend is observed in other eye cell types. This distinction between the eye, especially the retina, and the brain might be attributed to the eye’s constant exposure to light radiation. Another potential factor is the downregulation of ADAM17, the enzyme initiating Aβ’s non-amyloidogenic processing [
80]. PrP
C itself is believed to regulate ADAM17/TACE levels, indicating a mutual and intricate control mechanism [
81,
82,
83,
84,
85]. This proteolytic molecule is activated in immune cells, where TNF-α initiates several pathways crucial for their activity [
86]. Additionally, PrP
C stabilizes the extracellular matrix by interacting with β1 integrin [
87,
88]. When PrP
C expression is reduced, the RhoA-associated coiled-coil-containing kinase (ROCK) pathway is activated. Overactivation of ROCK, through the LIMK-cofilin pathway [
89,
90], results in a shift in the cytoskeleton status. These changes lead to increased resistance to aqueous outflow, elevated IOP, and ultimately, RGC loss.
In the brain, PrP
C acts as a radical scavenger. PrP
C-knockdown mice exhibit increased susceptibility to intracellular ROS [
91]. The N-terminal octapeptide repeat of PrP
C coordinates redox-active metals (e.g., Cu, Zn, and Fe) [
92,
93,
94,
95], enabling it to act as both a scavenger and a metal transporter [
81,
96,
97]. Given these findings, further research is needed to fully understand PrP’s multifaceted role in the eye [
4]. Striebel et al. [
32] and others have shown that prion-induced degeneration of photoreceptor cells in mice and humans resembles the pathology of human retinitis pigmentosa caused by retinal protein gene mutations. The Scrapie isoform of the prion protein (PrP
Sc) [
98] has been found to be associated with the base of cilia and swollen cone inner segments. These findings suggest that ciliopathy might be the underlying pathogenic mechanism. PrP
Sc has also been detected on the dendrites of cone and rod bipolar cells, extending into ribbon synapses. It is plausible that a similar mechanism might be activated in prion-like diseases, such as AD, as well as in human retinitis pigmentosa (RP) [
32].
Not only AD but also other prion and “prion-like” neurodegenerative progressions lead to retinal pathological changes in humans and other species. While there may be some overlap in histopathological abnormalities, each disease seems to have a unique impact on the retina [
99]. For instance, in patients with Parkinson’s disease (PD), abnormal α-synuclein oligomers [
100] have been observed in the retina, particularly in the nerve fiber (ganglion cell axons), inner plexiform, and ganglion cell body layers [
99].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11123258