Wet age-related macular degeneration (wAMD) is a chronic inflammation-associated neurodegenerative disease affecting the posterior part of the eye in the aging population. Aging results in the reduced functionality of cells and tissues, including the cells of the retina. Initiators of a chronic inflammatory and pathologic state in wAMD may be a result of the accumulation of inevitable metabolic injuries associated with the maintenance of tissue homeostasis from a young age to over 50. Apart from this, risk factors like smoking, genetic predisposition, and failure to repair the injuries that occur, alongside attempts to rescue the hypoxic outer retina may also contribute to the pathogenesis. Aging of the immune system (immunosenescence) and a compromised outer blood retinal barrier (BRB) result in the exposure of the privileged milieu of the retina to the systemic immune system, further increasing the severity of the disease.
- injury
- autoantigens
- protective autoimmunity
- inflammation
- aging
1. Introduction
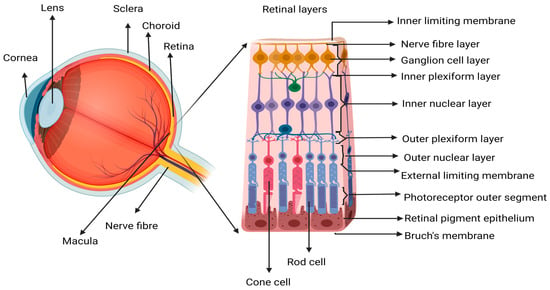
2. Inflammation, Immune Response, and Repair in Wet AMD
Immunosenescence
3. The Concept of Autoimmunity in AMD
| No | Autoantigens in AMD | References |
|---|---|---|
| 1 | Cardiolipin | [70] |
| 2 | Retinol binding protein (RBP3) | [9] |
| 3 | Aldolase C (ALDOC), | [9] |
| 4 | Retinaldehyde binding protein 1 (RLBP1) | [9] |
| 5 | Pyruvate kinase isozyme M2 (PKM2) | [9] |
| 6 | Carboxyethyl pyrrole (CEP) | [63] |
| 7 | Annexin A5 | [62] |
| 8 | HSPA8 | [62] |
| 9 | HSPA9 | [62] |
| 10 | HSPB4/CRYAA (Alpha-A Crystallin) | [62] |
| 11 | Protein S100-A9/calgranulin B | [62] |
| 12 | Alpha-enolase | [68] |
| 13 | Alpha-crystallin | [68][69] |
| 14 | Glial fibrillary acidic protein (GFAP) | [68] |
| 15 | CD5L/Apoptosis Inhibitor of macrophage (AIM) | [64] |
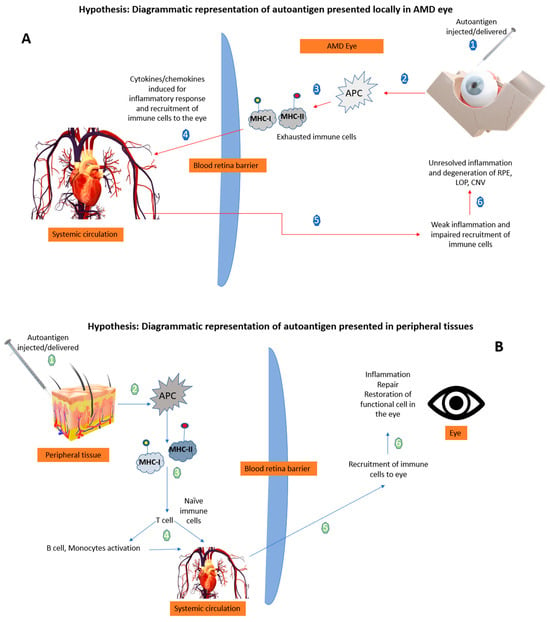
Therapeutic Delivery of Autoantigens

This entry is adapted from the peer-reviewed paper 10.3390/life13122236
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, E106–E116.
- WHO. World Report on Vision; WHO: Geneva, Switzerland, 2019.
- Hernández-Zimbrón, L.F.; Zamora-Alvarado, R.; La Paz, L.O.-D.; Velez-Montoya, R.; Zenteno, E.; Gulias-Cañizo, R.; Quiroz-Mercado, H.; Gonzalez-Salinas, R. Age-Related Macular Degeneration: New Paradigms for Treatment and Management of AMD. Oxid. Med. Cell. Longev. 2018, 2018, 8374647.
- Augustin, A.J.; Kirchhof, J. Inflammation and the pathogenesis of age-related macular degeneration. Expert Opin. Ther. Targets 2009, 13, 641–651.
- Gu, X.; Neric, N.J.; Crabb, J.S.; Crabb, J.W.; Bhattacharya, S.K.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related changes in the retinal pigment epithelium (RPE). PLoS ONE 2012, 7, e38673.
- Frank, R.N.; Puklin, J.E.; Stock, C.; Canter, L.A. Race, iris color and age-related macular degeneration. Trans. Am. Ophthalmol. Soc. 2000, 98, 109–115.
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 2000, 107, 2224–2232.
- Nussenblatt, R.B.; Lee, R.W.J.; Chew, E.; Wei, L.; Liu, B.; Sen, H.N.; Dick, A.D.; Ferris, F.L. Immune responses in age-related macular degeneration and a possible long-term therapeutic strategy for prevention. Am. J. Ophthalmol. 2014, 158, 5–11.e2.
- Morohoshi, K.; Ohbayashi, M.; Patel, N.; Chong, V.; Bird, A.C.; Ono, S.J. Identification of anti-retinal antibodies in patients with age-related macular degeneration. Exp. Mol. Pathol. 2012, 93, 193–199.
- Bonilha, V. Age and disease-related structural changes in the retinal pigment epithelium. Clin. Ophthalmol. 2008, 2, 413–424.
- Chirco, K.R.; Sohn, E.H.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye 2017, 31, 10–25.
- Hageman, D.A.G.S.; Gehrs, K.; Johnson, L.V. Age-related macular degeneration (AMD). Organ. Retin. Vis. Syst. 2009, 66, 1–44.
- Kauppinen, A.; Kaarniranta, K.; Salminen, A. Potential Role of Myeloid-Derived Suppressor Cells (MDSCs) in Age-Related Macular Degeneration (AMD). Front. Immunol. 2020, 11, 1–12.
- Philippidis, A. StockWatch: Apellis Wows Analysts with First FDA Approval for GA Therapy. Genet. Eng. Biotechnol. News 2023, 5, 186–191.
- Borchert, G.A.; Shamsnajafabadi, H.; Hu, M.L.; De Silva, S.R.; Downes, S.M.; MacLaren, R.E.; Xue, K.; Cehajic-Kapetanovic, J. The Role of Inflammation in Age-Related Macular Degeneration—Therapeutic Landscapes in Geographic Atrophy. Cells 2023, 12, 2092.
- Khan, M.; Aziz, A.A.; Shafi, N.A.; Abbas, T.; Khanani, A.M. Targeting Angiopoietin in Retinal Vascular Diseases: A Literature Review and Summary of Clinical Trials Involving Faricimab. Cells 2020, 9, 1869.
- Hussain, R.M.; Shaukat, B.A.; Ciulla, L.M.; Berrocal, A.M.; Sridhar, J. Vascular endothelial growth factor antagonists: Promising players in the treatment of neovascular age-related macular degeneration. Drug Des. Dev. Ther. 2021, 15, 2653–2665.
- Liberski, S.; Wichrowska, M.; Kocięcki, J. Aflibercept versus Faricimab in the Treatment of Neovascular Age-Related Macular Degeneration and Diabetic Macular Edema: A Review. Int. J. Mol. Sci. 2022, 23, 9424.
- Shirley, M. Faricimab: First Approval. Drugs 2022, 82, 825–830.
- Song, D.; Liu, P.; Shang, K.; Ma, Y.B. Application and mechanism of anti-VEGF drugs in age-related macular degeneration. Front. Bioeng. Biotechnol. 2022, 10, 943915.
- Van Bergen, T.; Etienne, I.; Cunningham, F.; Moons, L.; Schlingemann, R.O.; Feyen, J.H.M.; Stitt, A.W. The role of placental growth factor (PlGF) and its receptor system in retinal vascular diseases. Prog. Retin. Eye Res. 2019, 69, 116–136.
- Nair, A.A.; Finn, A.P.; Sternberg, P. Spotlight on Faricimab in the Treatment of Wet Age-Related Macular Degeneration: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2022, 16, 3395–3400.
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The role of inflammation in age-related macular degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001.
- Agarwal, M.; Majumder, P.D.; Babu, K.; Konana, V.; Goyal, M.; Touhami, S.; Stanescu-Segall, D.; Bodaghi, B. Drug-induced uveitis: A review. Indian J. Ophthalmol. 2020, 68, 1799–1807.
- Winkler, T.W.; Grassmann, F.; Brandl, C.; Kiel, C.; Günther, F.; Strunz, T.; Weidner, L.; Zimmermann, M.E.; Korb, C.A.; Poplawski, A.; et al. Genome-wide association meta-analysis for early age-related macular degeneration highlights novel loci and insights for advanced disease. BMC Med. Genom. 2020, 13, 120.
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112.
- Bradley, D.T.; Zipfel, P.F.; Hughes, A.E. Complement in age-related macular degeneration: A focus on function. Eye 2011, 25, 683–693.
- Detrick, B.; Hooks, J.J. The RPE Cell and the Immune System. In Retinal Pigment Epithelium in Health and Disease; Springer: Cham, Switzerland, 2020.
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786.
- Schwartz, M.; Shechter, R. Systemic inflammatory cells fight off neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 405–410.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725.
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852.
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882.
- Telander, D.G. Inflammation and age-related macular degeneration (AMD). Semin. Ophthalmol. 2011, 26, 192–197.
- Osuka, A.; Ogura, H.; Ueyama, M.; Shimazu, T.; Lederer, J.A. Immune response to traumatic injury: harmony and discordance of immune system homeostasis. Acute Med. Surg. 2014, 1, 63–69.
- Lazzara, F.; Conti, F.; Platania, C.B.M.; Eandi, C.M.; Drago, F.; Bucolo, C. Effects of Vitamin D3 and Meso-Zeaxanthin on Human Retinal Pigmented Epithelial Cells in Three Integrated in vitro Paradigms of Age-Related Macular Degeneration. Front. Pharmacol. 2021, 12, 778165.
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172.
- Ma, W.; Zhang, Y.; Gao, C.; Fariss, R.N.; Tam, J.; Wong, W.T. Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci. Rep. 2017, 7, 8433.
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22.
- Schwartz, M.; Raposo, C. Protective autoimmunity: A unifying model for the immune network involved in CNS repair. Neuroscientist 2014, 20, 343–358.
- Kent, D.; Sheridan, C. Choroidal neovascularization: A wound healing perspective. Mol. Vis. 2003, 9, 747–755.
- Kent, D.L. Age-related macular degeneration: Beyond anti-angiogenesis. Mol. Vis. 2014, 20, 46–55.
- Hünig, T.; Beyersdorf, N.; Kerkau, T. CD28 co-stimulation in T-cell homeostasis: A recent perspective. Immunotargets Ther. 2015, 2015, 111–122.
- Madelung, C.F.; Falk, M.K.; Sørensen, T.L. The association between neovascular age-related macular degeneration and regulatory T cells in peripheral blood. Clin. Ophthalmol. 2015, 9, 1147–1154.
- Faber, C.; Singh, A.; Falk, M.K.; Juel, H.B.; Sørensen, T.L.; Nissen, M.H. Age-related macular degeneration is associated with increased proportion of CD56+ T cells in peripheral blood. Ophthalmology 2013, 120, 2310–2316.
- Behnke, V.; Wolf, A.; Langmann, T. The role of lymphocytes and phagocytes in age-related macular degeneration (AMD). Cell. Mol. Life Sci. 2020, 77, 781–788.
- Boldison, J.; Chu, C.J.; Copland, D.A.; Lait, P.J.P.; Khera, T.K.; Dick, A.D.; Nicholson, L.B. Tissue-Resident Exhausted Effector Memory CD8 + T Cells Accumulate in the Retina during Chronic Experimental Autoimmune Uveoretinitis. J. Immunol. 2014, 192, 4541–4550.
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451.
- Ascunce, K.; Dhodapkar, R.M.; Huang, D.; Hafler, B.P. Innate immune biology in age-related macular degeneration. Front. Cell Dev. Biol. 2023, 11, 1118524.
- Voigt, V.; Wikstrom, M.E.; Kezic, J.M.; Schuster, I.S.; Fleming, P.; Makinen, K.; Daley, S.R.; Andoniou, C.E.; Degli-Esposti, M.A.; Forrester, J.V. Ocular antigen does not cause disease unless presented in the context of inflammation. Sci. Rep. 2017, 7, 14226.
- Adamus, G. Are anti-retinal autoantibodies a cause or a consequence of retinal degeneration in autoimmune retinopathies. Front. Immunol. 2018, 9, 765.
- Toker, A.; Slaney, C.Y.; Bäckström, B.T.; Harper, J.L. Glatiramer acetate treatment directly targets CD11b+Ly6G- monocytes and enhances the suppression of autoreactive T cells in experimental autoimmune encephalomyelitis. Scand. J. Immunol. 2011, 74, 235–243.
- Butovsky, O.; Koronyo-Hamaoui, M.; Kunis, G.; Ophir, E.; Landa, G.; Cohen, H.; Schwartz, M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA 2006, 103, 11784–11789.
- Avidan, H.; Kipnis, J.; Butovsky, O.; Caspi, R.R.; Schwartz, M. Vaccination with autoantigen protects against aggregated β-amyloid and glutamate toxicity by controlling microglia: Effect of CD4+CD25+ T cells. Eur. J. Immunol. 2004, 34, 3434–3445.
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870.
- Frederick, P.A.; Kleinman, M.E. The Immune System and AMD. Curr. Ophthalmol. Rep. 2014, 2, 14–19.
- Whitcup, S.M.; Sodhi, A.; Atkinson, J.P.; Holers, V.M.; Sinha, D.; Rohrer, B.; Dick, A.D. The role of the immune response in age-related macular degeneration. Int. J. Inflam. 2013, 2013, 348092.
- Adamus, G.; Chew, E.Y.; Ferris, F.L.; Klein, M.L. Prevalence of anti-retinal autoantibodies in different stages of Age-related macular degeneration. BMC Ophthalmol. 2014, 14, 1–9.
- Patel, N.; Ohbayashi, M.; Nugent, A.K.; Ramchand, K.; Toda, M.; Chau, K.Y.; Bunce, C.; Webster, A.; Bird, A.C.; Ono, S.J.; et al. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005, 115, 422–430.
- Morohoshi, K.; Patel, N.; Ohbayashi, M.; Chong, V.; Grossniklaus, H.E.; Bird, A.C.; Ono, S.J. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Exp. Mol. Pathol. 2012, 92, 64–73.
- Iannaccone, A.; Giorgianni, F.; New, D.D.; Hollingsworth, T.J.; Umfress, A.; Alhatem, A.H.; Neeli, I.; Lenchik, N.I.; Jennings, B.J.; Calzada, J.I.; et al. Circulating autoantibodies in age-related macular degeneration recognize human macular tissue antigens implicated in autophagy, immunomodulation, and protection from oxidative stress and apoptosis. PLoS ONE 2015, 10, 1–22.
- Gu, X.; Meer, S.G.; Miyagi, M.; Rayborn, M.E.; Hollyfield, J.G.; Crabb, J.W.; Salomon, R.G. Carboxyethylpyrrole Protein Adducts and Autoantibodies, Biomarkers for Age-related Macular Degeneration. J. Biol. Chem. 2003, 278, 42027–42035.
- Iannaccone, A.; Hollingsworth, T.J.; Koirala, D.; New, D.D.; Lenchik, N.I.; Beranova-Giorgianni, S.; Gerling, I.C.; Radic, M.Z.; Giorgianni, F. Retinal pigment epithelium and microglia express the CD5 antigen-like protein, a novel autoantigen in age-related macular degeneration. Exp. Eye Res. 2017, 155, 64–74.
- Umeda, S.; Suzuki, M.T.; Okamoto, H.; Ono, F.; Mizota, A.; Terao, K.; Yoshikawa, Y.; Tanaka, Y.; Iwata, T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis). FASEB J. 2005, 19, 1581–1760.
- Morohoshi, K.; Goodwin, A.M.; Ohbayashi, M.; Ono, S.J. Autoimmunity in retinal degeneration: Autoimmune retinopathy and age-related macular degeneration. J. Autoimmun. 2009, 33, 247–254.
- Kubicka-Trzäska, A.; Wilańska, J.; Romanowska-Dixon, B.; Sanak, M. Circulating antiretinal antibodies predict the outcome of anti-VEGF therapy in patients with exudative age-related macular degeneration. Acta Ophthalmol. 2012, 90, e21–e24.
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Analysis of IgG antibody patterns against retinal antigens and antibodies to α-crystallin, GFAP, and α-enolase in sera of patients with “wet” age-related macular degeneration. Graefe Arch. Clin. Exp. Ophthalmol. 2007, 245, 619.
- Nakata, K.; Crabb, J.W.; Hollyfield, J.G. Crystallin distribution in Bruch’s membrane-choroid complex from AMD and age-matched donor eyes. Exp. Eye Res. 2005, 80, 821–826.
- Özkan, B.; Karabaş, L.V.; Altintaş, Ö.; Tamer, G.S.; Yüksel, N.; Ça, Y. Plasma antiphospholipid antibody levels in age-related macular degeneration. Can. J. Ophthalmol. 2012, 47, 264–268.
- Patel, P.B.; Shastri, D.H.; Shelat, P.K.; Shukla, A.K. Ophthalmic drug delivery system: Challenges and approaches. Syst. Rev. Pharm. 2010, 1, 113.
- Melo, G.B.; da Cruz, N.F.S.; Emerson, G.G.; Rezende, F.A.; Meyer, C.H.; Uchiyama, S.; Carpenter, J.; Shiroma, H.F.; Farah, M.E.; Maia, M.; et al. Critical analysis of techniques and materials used in devices, syringes, and needles used for intravitreal injections. Prog. Retin. Eye Res. 2021, 80, 100862.
- Falavarjani, K.G.; Nguyen, Q.D. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: A review of literature. Eye 2013, 27, 787–794.
- Bakalash, S.; Shlomo, G.B.; Aloni, E.; Shaked, I.; Wheeler, L.; Ofri, R.; Schwartz, M. T-cell-based vaccination for morphological and functional neuroprotection in a rat model of chronically elevated intraocular pressure. J. Mol. Med. 2005, 83, 904–916.
- Belokopytov, M.; Ben-Shlomo, G.; Rosner, M.; Belkin, M.; Dubinski, G.; Epstein, Y.; Ofri, R. Functional efficacy of glatiramer acetate treatment for laser-induced retinal damage in rats. Lasers Surg. Med. 2008, 40, 196–201.