Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Rheumatology
Rheumatoid arthritis (RA) is a chronic autoimmune disorder that has a significant impact on quality of life and work capacity. Treatment of RA aims to control inflammation and alleviate pain; however, achieving remission with minimal toxicity is frequently not possible with the current suite of drugs.
- rheumatoid arthritis
- drug repurposing
- connectivity mapping
1. Introduction
Rheumatoid arthritis (RA) is a systemic and chronic autoimmune disorder that affects musculoskeletal joints, resulting in persistent synovitis, hyperplasia, autoantibody production, cartilage and joint destruction, erosion, and functional impairment [1]. Extra-articular involvement of other organs in RA frequently results in dermatological, neurological, cardiovascular, pulmonary, renal, and gastrointestinal pathology [2].
RA has a global prevalence of 0.24–1%. It has a prevalence rate of approximately 1% in the UK, with an incidence of 1.5 and 3.6 per 100,000 in men and women, respectively, indicating a predilection towards women [3,4]. It is estimated that over 450,000 adults in the UK have rheumatoid arthritis [5]. The disorder can occur at any age, but the average age of onset is between 30 and 50 years [6].
Multiple genetic factors interacting with the environment have been implicated in the susceptibility to and pathogenesis of RA. Alleles of the highly polymorphic human leukocyte antigen (HLA) gene, particularly the HLA-DRB1 gene, have been associated with an increased risk of developing the disorder [7]. The presence of specific HLA-DRB1 alleles containing the shared epitope is known to contribute to the aberrant immune response and the production of autoantibodies, including rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) [8]. Genome-wide association studies have identified many non-HLA loci associated with RA susceptibility. Several well-established gene associations are PTPN22, CTLA4, STAT4 and PADI4 [7]. Polymorphism in the PTPN22 gene is one of the strongest associations of a non-HLA gene to the development of the disorder thought to influence the immune activation threshold of T cells and B cells [9]. CTLA4 genetic polymorphisms may influence T-cell activation and the balance between regulatory T cells and effector T cells, thus affecting immune tolerance [10]. STAT4 is involved in the regulation and promotion of pro-inflammatory cytokines, such as IL-12, IL-23 and type 1 interferon, which contribute to chronic inflammation in RA [11]. Studies have found that PADI4 gene variants contribute to the production of ACPA, leading to the progression of joint inflammation and destruction [12]. Environmental risk factors influencing the development of RA include smoking, air pollution, obesity, occupational exposures such as silica and textile dust, infections, vitamin D deficiency, immunisations, oral contraceptives, and socioeconomic status [7,13,14,15,16,17]. Epigenetic factors are understood to play a pivotal role in the pathogenesis of RA, contributing to the dysregulation of gene expression observed in this autoimmune disorder. DNA methylation alters gene silencing patterns, impacting immune regulation in RA. Histone modifications, like acetylation, methylation and citrullination, affect chromatin structure and gene transcription, increasing expression of genes linked to inflammation and autoimmunity [18]. The dysregulation of non-coding RNAs, particularly microRNAs (miRNAs), exert post-transcriptional control over gene expression, influencing immune response and contributing to joint damage.
2. Connectivity Mapping
Connectivity mapping (CMap) is a bioinformatic approach pioneered by Lamb et al. in 2006 with the basic concept of comparing a reference database of drug-related gene expression profiles with a query gene signature specific to a disease or a response to treatment in a disease [52]. This allows for the identification of associations between drugs and disease-related genes with the ultimate aim of predicting potential therapeutic options effective in that disease. Applications of CMap in pharmacogenomics include the discovery of novel phenotypic relations, elucidation of drug mechanism of action, drug repurposing and identification of drug combinations [52].
CMapBatch is a parallel approach to connectivity mapping adapted by Fortney et al. [53]. This approach is similar to meta-analysis as it applies CMAP to multiple gene signatures for the same disease and then combines the resulting outcomes [53]. Analysis of lung cancer data revealed that CMapBatch produces a more stable list of drugs when compared to individual gene signatures. Despite the fact that CMapBatch was only tested for lung cancer, the proposed meta-analysis can be used for any disease phenotype to prioritise therapeutics. For example, multiple colorectal cancer datasets were analysed to compile a gene signature consisting of 148 genes. CMap analysis with this signature identified 10 candidate compounds, including existing chemotherapies such as irinotecan and etoposide [54,55]. Other studies utilising CMap show promise by identifying candidate compounds and combination therapies for the treatment of breast cancer [56] and gastric cancer [57].
The CMap approach has been utilised for putative drug target investigations in autoimmune diseases. A study on Hashimoto’s thyroiditis performed CMap analysis on a human thyroid microarray dataset and found a causal link between viral infection and triggering or exacerbating the autoimmune response in the thyroid gland [58]. Comparisons of the disease gene signature against a perturbation gene expression database revealed potential markers and candidate drugs as promising therapeutics for the condition. Another study in multiple sclerosis, a complex inflammatory disease involving multiple disease pathways, used the CMap approach to analyse immune cell changes in transcriptomic datasets to identify potential target genes and candidate drugs from the CMAP database and DrugBank database that can be repositioned to engage multiple treatment pathways [59]. Cystic fibrosis and Huntington’s disease studies have validated the effectiveness of the CMap approach to identify small molecules with the potential to inhibit the disease state or regulate the expression of a small number of genes. For instance, A20 was identified as a key target to downregulate the pro-inflammatory NF-kB pathway, and the connectivity mapping approach predicted ikarugamycin and quercetin, FDA-approved drugs with anti-inflammatory effects, to induce A20 expression and therefore reduce the inflammatory response in cystic fibrosis [55]. Deferoxamine and chlorzoxazone, FDA-approved antioxidant and anti-inflammatory agents, were identified to reduce mutant HTT toxicity and HTT-induced caspase activation in PC12 cells, which can delay the onset or progression of Huntington’s disease [60].
3. Drug Repurposing and Sensitisation
Drug repurposing is a concept that has attracted considerable attention in recent years. The term drug repurposing is broadly defined as investigating drugs which are already approved for specific disease indications but may have utility in alternate diseases. The established safety profile of such drugs is a significant advantage, in addition to bypassing the time and cost involved with the de novo development of new compounds.
Monoclonal antibody treatments such as tocilizumab and mavrilimumab have been repurposed for use in COVID-19 and have been associated with reducing the incidence of severe infections and decreasing the duration of vasopressor support needed in severe patients. Studies on mavrilimumab concluded it was associated with improved clinical outcomes for severe COVID-19 patients with systemic hyperinflammation and pneumonia. JAK inhibitor, baricitinib, speeds up viral clearance and augments patients’ discharge rates compared to COVID-19 patients who have standard-of-care. A recent randomised clinical trial with 1033 patients showed a better therapeutic outcome of combined therapy of baricitinib with remdesivir for COVID-19 hospitalised patients compared to only remdesivir [61]. This highlights that the majority of recent research conducted in drug repurposing has focused on finding drugs that could combat the effects of the SARS-CoV-2 virus infection. Therefore, there is now a unique opportunity to apply similar principles in RA, a chronic and frequent treatment-refractory disease, to identify effective treatments which reduce disease activity and disease burden. On the other hand, the principle of drug sensitisation is when a drug exhibits synergistic effects with another drug to produce enhanced anti-disease efficacy that could not be achieved by using either drug in isolation [62]. The rationale behind this combined therapeutic approach is to target more than one disease-associated pathway during treatment. Such an approach is built on the premise that combination therapies simultaneously engage multiple pathways to evoke a higher response than those achieved with monotherapy. Another suggestion is that treatment with one drug can evoke a dynamic response, resulting in sensitivity to treatment with a second drug. It is believed that combinations of repurposed already approved drugs have good potential to achieve greater efficacy at lower dosages and may overcome drug resistance [63]. The implementation of synergistic combination therapy can raise concerns about synergistic toxicity as a result of targets and molecular mechanisms being shared between combined drugs. Rheumatology has widely adopted the concept of combination therapies, leading to improved outcomes in many cases [64]. However, there are multiple studies evidencing that drug combinations elevate the risk of adverse drug reactions.
There are still many drugs routinely used for the treatment of RA which are yet to have their combinational effects fully explored.
4. Bioinformatics Pipelines to Identify Potential Therapeutics
Bioinformatic-led approaches are now more widely implemented within drug discovery pipelines for immune-mediated and inflammatory diseases. Recent studies illustrate the potential of bioinformatic approaches to exploit increasing volumes of data generated from clinical trials and studies carried out globally. Bioinformatic methods have been used to create data warehouses, algorithms, networks, and programs to analyse “big data” [67]. Drug development pipelines using bioinformatic resources and techniques have strong potential to accelerate candidate identification, avoid unwanted side effects and predict drug resistance [68].
For example, a drug discovery strategy was developed to identify potential therapeutic agents for inflammatory bowel disease. Data involving the NF-κB/RelA pathways were curated from multiple sources, including sequencing data, text-mining of relevant abstracts, genome-wide association studies and HumanPSD database [69]. Potential target genes within the pathways were classified as master regulators for pathway analysis. Prediction of activity spectra was used to assess the association between the chemical structure of compounds and their biological activities to identify potential novel drugs for inflammatory bowel disease treatment. Results of the study indicated that clarithromycin, a macrolide antibiotic, has the potential to act as an inhibitor of the NF-κB signalling pathway in the gastrointestinal tract. This finding complements existing clinical literature, as macrolides are already used to treat inflammatory conditions, such as panbronchiolitis [70] and atopic dermatitis [71]. The antibacterial and immunomodulatory properties of macrolides have shown promise in inhibiting the production and secretion of pro-inflammatory cytokines [71]. Additional studies investigating the effect of macrolide in combination with rifabutin for the treatment of Crohn’s disease indicate significant improvement in patient outcomes and disease activity [72]. This drug discovery approach incorporated an intentional bias towards target genes involved in the NF-κB signalling pathways, which resulted in corticosteroids and NSAIDs as the majority of predicted drugs.
An integrative computational modelling approach was developed to identify effective therapeutic agents for CD4+ T cell-mediated immune disorders. Multi-omic data was used to construct genome-scale metabolic models of CD4+ T cells to show perturbation in rheumatoid arthritis, multiple sclerosis, and primary biliary cholangitis. In silico simulations were performed on these models to predict drug targets from existing FDA-approved drugs and compounds with the potential to downregulate effector CD4+ T cells. Sixty-eight potential drug targets were identified and validated in vitro to propose several drugs that can be repurposed for RA, multiple sclerosis, and primary biliary cholangitis treatment [73].
5. Application of Artificial Intelligence in RA
Drug repurposing and use of artificial intelligence (AI) to accelerate discovery in legacy data is a concept that has garnered growing interest from pharmaceutical companies and research organisations in recent years, with several RA focussed studies proposing a computation-based drug discovery approach. One such study integrated drug-related and disease-related data to construct a genetic disease network to develop a drug-ranking algorithm. This algorithm discovered innovative drugs from diseases genetically related to RA that can be repositioned to treat RA [74]. A preclinical study demonstrated that repurposing of pirfenidone, a drug originally used to treat anti-pulmonary fibrosis, can inhibit inflammation and angiogenesis via multiple pathways in collagen-induced arthritic rats. This finding supports literature proposing the use of pirfenidone in RA; however, it requires further study in humans to reveal the potential of being used as a therapeutic in rheumatoid arthritis [75].
A further study used bioinformatic approaches to establish a transcriptional regulatory network to identify tissue-specific repurposing drug candidates for RA. The candidate drugs were reviewed and ranked based on supporting evidence obtained from extensive literature searches using text-mining analyses. Momelotinib, ibrutinib, and sodium butyrate were suggested as promising drug candidates, but further clinical studies are required to fully elucidate their therapeutic effects in patients with RA [76].
Medical image data also play a crucial role in understanding disease mechanism, progression and severity in RA. While image data is not commonly used as the primary input for AI-based drug discovery in RA, in recent years, data from various imaging techniques, such as MRI and ultrasound scans, have been used to develop algorithms and models to quantify synovitis and assess severity [75,77,78].
Numerous studies analysing treatment regimens in RA show that double and triple therapy leads to greater clinical outcomes than DMARD monotherapy. Combination therapy administered early in the course of the disease has been found to significantly decrease disease activity [79]. This finding aligns with the rationale behind the concept of drug sensitisation that administering multiple drugs in combination engages multiple pathways to evoke a higher treatment response. However, current evidence on combination therapy is limited, with knowledge gaps that can only be filled with further research and randomised controlled trials of adequate power.
The above studies demonstrate the power of implementing an in silico drug discovery model to identify repurposed candidate drugs and highlight the importance of incorporating steps to orthogonally validate results to determine which drugs to pursue for further experimental investigations.
As an exemplar approach, we developed a novel bioinformatic pipeline (Figure 3), DrugExpress, which integrates the connectivity mapping platform sscMap (statistically significant connectivity map) [80] and ZhangScore [81,82]. DrugExpress will identify drug combinations, dosage regimens and already FDA-approved drugs that can be repurposed in rheumatoid arthritis. This pipeline also incorporates the novel concept of drug sensitisation by predicting drugs that will act as a sensitiser to another drug to produce synergistic effects which enhance therapeutic efficacy by targeting multiple disease pathways. Suitable candidate drugs will be identified and shortlisted based on their abilities to shift the transcriptomic (gene expression) profiles of treatment-naïve disease and to sensitise non-responding patient sub-groups towards more favourable “response-like” transcriptome profiles. Candidate drugs subsequently undergo toxicity screening and pathway analysis. The final list of candidate drugs then requires validation in vitro in RA model systems.
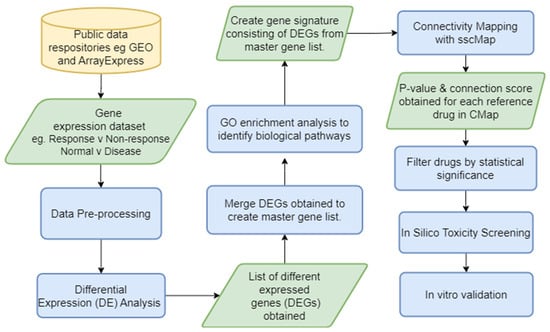
Figure 3. DrugExpress pipeline—In silico drug discovery and repurposing pipeline with an in vitro validation endpoint. GEO—Gene Expression Omnibus, DE—differential expression, DEG—differentially expressed genes, GO—gene ontology, sscMap—statistically significant connectivity map. Yellow cylinder represents the start point of the pipeline, green parallelogram represents input/output of a process and blue rectangle represents a process.
Figure 4 shows results from expression data after interrogation using the DrugExpress pipeline. Public datasets such as the Gene Expression Omnibus and ArrayExpress are mined to gather a collection of suitable datasets and pre-processed manually using Microsoft Excel and R programming. The datasets are filtered, sorted and selected based on the presence of disease activity scores, availability of clinical features, sample count and technological platform. Differential expression analysis was performed comparing response and non-response to treatment to obtain a list of differentially expressed genes (DEGs) characteristic of treatment response. DEGs from multiple datasets are merged to create a master gene list and subsequently mapped to Affymetrix probeset IDs to create a treatment response gene signature. Connectivity mapping (CMap) analysis is used to establish networks between DEGs in the response gene signature and FDA-approved drugs. p-value and connection scores of each reference drug in the CMap were obtained and used to determine statistical significance and perturbation stability. A total of six statistically significant candidate compounds were identified with a high probability of inducing therapeutic response. The next step is to perform in silico toxicity screening on the list of candidate compounds ahead of in vitro verification on optimal compounds to assess efficacy and ability to reduce gene expression in key pathways and cell proliferation associated with lower disease activity in MH7A human Ra synovial fibroblasts. This illustrates how publicly available expression datasets can be used to predict the theoretical effect of drug candidates and prioritise novel compounds with maximal potential to reduce disease activity.
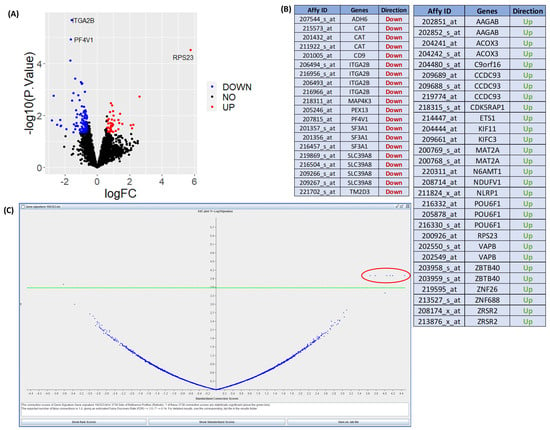
Figure 4. Results using the DrugExpress pipeline. (A) Volcano plot showing differentially expressed genes (labelled) between responders and non-responders to combined treatment of sulfasalazine and methotrexate from one suitable dataset. (B) Table of merged list of differentially expressed genes from multiple datasets. (C) Volcano plot of statistically significant candidate compounds located above the green threshold line with a perturbation stability score of 1. Statistically significant candidate drugs which would induce theoretical reduction in disease activity are located in the circled area of plot.
This entry is adapted from the peer-reviewed paper 10.3390/jpm13121633
This entry is offline, you can click here to edit this entry!