Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Plant Sciences
A genome is an organism’s comprehensive collection of nucleic acids (DNA or RNA), which contains all of its genes. Genomic science or genomics is the study of the genome’s structure, function, evolution, mapping and modifications.
- molecular
- DNA
- PCR
- plant
1. Introduction
Recent breakthroughs in molecular biological techniques have accelerated the pace of high-throughput genome sequencing, genomic characterization and gene expression analysis [27]. The technique of decoding the genome using high throughput next-generation sequencing (NGS) technology comprises the isolation of genomic DNA, the multiplication of DNA using polymerase chain reaction (PCR), the sequencing of the DNA and the assessment of the sequence’s integrity [28]. The sequencing and assembly of DNA, followed by the structural and functional annotation of the gene, permits large-scale investigations into the activities of genes and elucidates the interactions of gene products at the cellular and organismal levels [29]. The field of genomics has been discussed under three categories in the following sections (Figure 1):
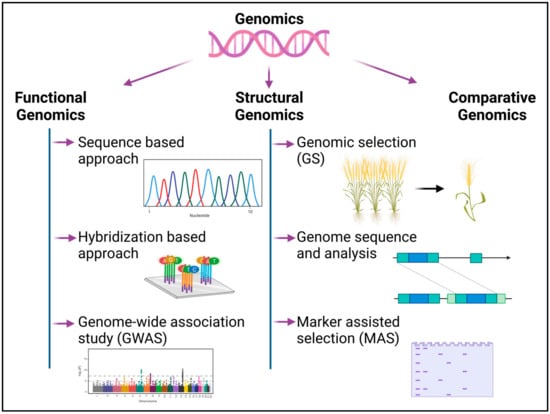
Figure 1. Different cohorts of genomics for crop assessment and improvement in relation to abiotic-stress tolerance response. The diagram was created using BioRender (https://biorender.com/) premium version.
2. Functional Genomics
Functional genomics analyzes the data generated by complete or partial genome sequencing to describe gene functions and interactions and employs two complementary approaches to the determination of individual genes, viz., forward and reverse genetics [30]. The forward genetic approach investigates a randomly obtained mutant of an interesting phenotype and identifies the responsible gene(s). On the other hand, the reverse genetic approach is the analysis of an organism’s phenotype by disruption of a known gene [31]. The technique of functional genomics helps in unravelling the gene interactions as well as the regulatory networks of genes and this technique employs the below-mentioned methodologies:
2.1. Sequencing-Based Approaches
Exploring the expressed gene catalogue has been possible by analyzing the ESTs, i.e., the gene sequence produced from the cDNA clones by the single-pass method [32]. Utilizing the ESTs is a cost-effective as well as rapid method and is thus considered mainly in functional genomics studies. Deokar et al. [33] conducted an EST-based investigation in which they found differentially expressed genes (DEGs) in drought-susceptible and tolerant plants using the suppression subtraction hybridization (SSH) approach to build the analyzed plant’s EST library. After obtaining the ESTs, it gets submitted to the National Center for Biotechnological Information (NCBI), which serves as the source for EST sequencing to reveal the genes that are differentially expressed. Another sequence-based approach is Serial Analysis of Gene Expression (SAGE) which helps in quantifying the abundance of several transcripts together. In the SAGE, sequence tags of small stretch are joined and thereafter sequenced to analyze the gene expression [34] and the identification by these short tags depends on the presence of the EST database for a given species of consideration. SAGE technique is not very applicable to plant systems and thus has been modified as either SuperSAGE or DeepSAGE [35]. Similar to SAGE, the Massively Parallel Signature Sequencing (MPSS) approach has been used to study the long sequences with tags that are affixed to microbeads and then sequenced in parallel, allowing for the analysis of millions of transcripts simultaneously [36]. MPSS technique has high throughput and thus enables identifications to be performed with more specificity.
2.2. Hybridization-Based Approaches
Another approach to studying the sequence is an array-based technique, where the hybridization of the DNA that has to be studied is carried out with the cDNA/oligonucleotide probes to assess the gene expression [37]. The limitation of this approach is that designing the probe requires the knowledge of the transcript either in the form of a sequence or a clone. Array-based data exist extensively for model plant species but there is a lack of data for the economically important crop plants and thus unravelling the stress responses utilizing these methods in crop plants becomes a difficult task.
2.3. Expansions to Functional Genomics Approaches
Genome-wide association studies (GWAS) are an experimental and statistical examination of a large number of genetic variations across the genome in different organisms (or individuals) to determine whether any variant is related to a trait of interest. GWAS examines the entire genome to identify DNA variations related to the trait of interest [38]. GWAS has been successfully used in deciphering abiotic tolerance in rice [39], soybean [40], wheat [41], maize [42], sesame [43], barley [44], chickpea [45], rapeseed [46], cotton [47,48,49] and sorghum [50,51]. The primary goal of GWAS is to identify genomic areas linked with agronomic or morphological features or any phenotypes that can be markers, genes or quantitative trait loci (QTL) for gene discovery, introgressive hybridization and MAB [52] (Table 1). Advances in genomics and phenomics have resulted in a more precise and comprehensive characterization of QTLs, often referred to as QTLomes [53]. Presently, the QTLome concept is being utilized in specific QTL alleles associated with traits. In addition, numerous statistical methods, such as meta-QTL analysis, have aided in the collection of QTL data from various studies on the same linkage to pinpoint the precise QTL region [41]. This meta-analysis has been applied to study important crop plants such as wheat, soybean, etc. Utilizing the meta-analysis study, Ha et al. [54] identified loci for salt tolerance in soybean on chromosome 3 and used simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers to analyze the RIL population (PI 483463 Hutcheson). Sheoran et al. [55] identify the candidate genes of maize for abiotic stress tolerance and utilization in future breeding for crop improvement. Moreover, such meta-QTL analysis also helped to screen the genomic loci of rice for salinity and drought tolerance in different growth and developmental phases—seedling and flowering stages [56,57].
In the 21st century, gene editing has emerged very alarmingly for functional characterization and validation of newly identified genes or genetic regions associated with stress-responsive genes in plants [58]. Success in manipulating a specific gene with a respective function may be achieved by the use of the clustered regularly interspaced short palindromic repeat (CRISPR)—Cas (CRISPR-associated system), which is a more concise, less labor-intensive alternative to traditional methods such as meganucleases (MNs), zinc-finger nucleases (ZFNs), transcription-activator-like effector nucleases (TALENs) [59]. The CRISPR-Cas system of gene editing approach becomes very efficient to characterize the functionality of plant-responsive genes for drought [60,61], salinity [62,63], heat [64] and cold stress [65].
3. Structural Genomics
Functional genomics is concerned with the function of genes and their interactions. Structural genomics is concerned with determining the three-dimensional structure of genes to identify, locate and determine their order along the chromosome [60]. Functional and structural genomics studies corroborate the intricate links between sequence and structure, ultimately offering the complete genome, which can aid in the understanding of a wide range of biological issues [66].
3.1. Genomic Selection (GS)
Genomic selection (GS) permits the quick selection of better genotypes by utilizing high-density markers distributed across the genome [67]. GS is a novel strategy for optimizing quantitative characteristics; it utilizes marker and phenotypic data from observed populations to assess the impact of all loci [68,69]. Generally, the method of genomic selection relies on two types of datasets: a training set and a validation set [70,71]. The training data set is the reference population and is used for the estimation of marker effects, whereas the validation set possesses the selected candidates that have been genotyped [72].
3.2. Genome Sequencing and Mapping
DNA sequencing has provided many details on the sequence, including the whole genome. There are several platforms, such as Roche 454GS FLX Titanium or Illumina Solexa Genome Analyzer, that are said to be NGS platforms that have helped in reducing the sequencing cost as well as time in comparison to conventional sequencing methods such as the Sanger method [73]. The sequencing method helps in developing improved varieties of crops by sequencing and resequencing processes. To date, the genomic sequence for several crop plants such as rice, wheat, maize, sorghum, soybean and tomato has been published. Apart from these crop plants, the sequence of model plants such as Arabidopsis thaliana and Brachypodium distachyon has also been published [74]. Genome sequencing provides detailed data on the features of genomes (coding as well as non-coding genes), GC content, repetitive elements as well as regulatory sequences [75]. Although genome sequencing provides important details for improving crops using molecular breeding, its usefulness is limited to species that have a smaller genome. To facilitate a complex genome study, another technology that is chromosome-specific has helped in developing Bacterial Artificial Chromosome (BAC) libraries to help in studying the complex genome. Mapping of 1 Gb chromosome of wheat has been possible with the help of the chromosome-by-chromosome approach only [76]. Mapping compiles genetic mappings into physical contigs as well as providing a framework for the assembly of sequences into the whole genome, and in the absence of a reference genome sequence, this BAC-end shotgun sequence gives details of genome evolution as well as structure [77,78]. Another interesting method for developing a whole genome sequence has been carried out by detecting the QTL using a methodology named QTL-seq. A QTL is a polymorphic locus that differentially affects the trait. QTL mapping is the technique of utilizing DNA markers to generate linkage maps and identify genomic areas linked with certain characteristics. QTL mapping is used to characterize the organization and evolution of the chromosomes [79,80]. So far, several QTLs have been reported for tolerance to drought [81,82], salinity [39,83,84,85,86], heat [87,88], cold [46,89], etc. (Table 1).
Table 1. Important QTLs/markers identified for abiotic stress response in field crops.
| Plants | QTLs/Markers | Chr. Location | Methods Used | Abiotic Stresses | References |
|---|---|---|---|---|---|
| Rice | OsHKT1;1 | Chr 1 | GWAS | Salinity | [39] |
| qWUE.STI6 | Chr 6 | Linkage mapping | Drought | [80] | |
| Saltol | Chr 1 | Linkage mapping | Salinity | [87] | |
| qCTBB2 qCTBB3 |
Chr 2 Chr 3 |
Linkage mapping | Cold | [89] | |
| qSTS4 | Chr 4 | QTL-seq | Salinity | [90] | |
| Soybean | AX-93897192 | Chr 19 | GWAS | Phosphorus efficiency | [40] |
| qGI10-1 | Chr 10 | GWAS | Drought | [79] | |
| qSFT_3-38, qSFT_7-3 |
Chr 3 Chr 7 |
Linkage mapping | Flooding | [91] | |
| qST6 qST10 |
Chr 6 Chr 10 |
Genotype-based sequencing (GBS) | Salinity | [92] | |
| Wheat | MQTL1D.4 MQTL2D.5 MQTL3A.1 |
Chr 1D Chr 2D Chr 3A |
MetaQTL | Drought stress Heat, Salinity Waterlogging |
[41] |
| QNa.asl-2A | Chr 2A | Genotype-based sequencing (GBS) | Salinity | [81] | |
| YIELD_MQTL4B.2_D | Chr 4B | MetaQTL | Heat, Drought | [85] | |
| qWMs108_7-1 | Chr 7-3A | Linkage mapping | Drought | [93] | |
| QSpad3.ua-1D.5 | Chr 1D | GWAS | Waterlogging | [94] | |
| QMrl3B(T2|T1) | Chr 3B | Linkage mapping | Salinity | [95] | |
| Maize | Zm00001eb013650 | Chr 1-10 | GWAS + RNAseq | Salinity | [42] |
| qPOD2b | Chr 2 | Genome-Wide Association Study (GWAS) | Cold | [96] | |
| Rapeseed | SA07_23415428 | Chr SA07 | GWAS | Freezing | [45] |
| qDSI_SL-11-3 qDSI_RL-11-1 qDSI_RL-11-4 qDSI_SL11-3 | Chr C01 | Linkage mapping | Drought, Freezing | [97] | |
| qRRL.A3b | Chr A03 | Linkage mapping | Waterlogging | [98] | |
| Barley | QcRWC.3H_2.1 QcWC.3H_1 3H |
Chr 3H | Linkage mapping | Drought | [76] |
| HORVU2Hr1G111780.3 | Chr 2H | Linkage mapping | Salinity | [82] | |
| qSLS-4 | Chr 4H | Linkage mapping | Salinity | [88] | |
| QBIO.2H | Chr 2H | GWAS | Waterlogging | [99] | |
| Cotton | qtlCSI01 | Chr 3 | Composite interval mapping | Drought | [47] |
| qGR-Chr4-3, qFER-Chr12-3, qFER-Chr15-1 | Chr 4 Chr 12 Chr 15 |
Linkage mapping | Salinity | [48] | |
| qEC_A02_ck qFW_A06_150.1 |
Chr 2 Chr 6 |
Genotyping by Sequencing (GBS) | Salinity | [49] | |
| qFSHa1 | Chr 15 | Composite interval mapping | Heat | [86] | |
| Sorghum | qPH-6 qMC2-9 |
Chr 6 Chr 9 |
Genotype-based sequencing (GBS) | Excess soil nitrogen | [50] |
| qTB45_4.S | Chr 4 | Linkage mapping | Salinity | [51] |
3.3. Molecular Marker Resources
DNA markers are short areas of DNA sequences that have the ability to identify variations in a population’s DNA or polymorphisms (base deletion, insertion and substitution), including base deletions, insertions and substitutions. DNA markers are also known as genetic markers [11]. Molecular markers aid in tagging genomic traits such as pathogen resistance, abiotic stress tolerance, quantitative analysis, etc. Recent advancement in this resource has provided a new horizon for the genetic improvement of traits for stresses such as drought, salt, etc. [11]. To date, several molecular markers have been reported that help in identifying polymorphism in plants, and these markers include random amplified polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), SNP, SSR and sequence-tagged sites (STS) [11,100]. Restriction fragment length polymorphism (RFLP) is the most basic marker that helps in identifying the polymorphism arising due to mutation or deletion/insertion leading to either formation/deletion of endonuclease recognition sites in restriction fragment length [101]. Another marker, RAPD, which is generated via random primers, identifies complementary sites at a short distance within the genome, while AFLP combines the restriction digestion as well as the PCR amplification and thus helps in identifying the linkages [102]. The SSR or microsatellite markers are tandem repeats of short mono-, di-, tri- and tetra-nucleotides and help in measuring the genetic diversity among species and also differentiate alleles that are homozygotic and heterozygotic between the lines from the same origin [103]. The SNPs are used for the characterization of germplasm as well as gene mapping. Due to their high abundance, codominance and sequence tagging they help in understanding complex traits utilizing microarrays such as Affymetrix GeneChip. Marker-assisted selection (MAS) is a genomic approach to identifying and breeding associated allelic markers [104]. During the process of marker-assisted selection, a characteristic of interest is chosen on the basis of a marker that has been associated with a particular or multiple abiotic stress [105,106]. Previously, success in MAS for abiotic stress tolerance was lagging due to the limited availability of genomic data. Genome databases and datasets that are very valuable in the construction of SSRs and SNP markers have been produced thanks to recent improvements in high-throughput DNA sequencing and genotyping technology [107]. Such availability of various high-throughput molecular markers and genome sequencing technologies leads to genomics-assisted breeding [108] and SNP difference-based haplotype mapping [109] to improve crops with stress tolerance properties.
4. Comparative Genomics
Comparative genomics is the science of comparing entire genomes or parts of genomes to find out basic biological similarities and differences as well as investigating evolutionary relationships between organisms [110]. Comparative genomics compares biological sequences by aligning them and detecting conserved sequences. Thus, studies of comparative genomics have revealed considerable synteny in related species [111]. Moreover, as comparative genomics can detect small-scale changes within different genomes, comparative studies of protein-coding regions and their consequences on protein structure and function identify important regulatory elements within DNA [112]. Comparative genomics gave rise to the “genome zipper” concept that helps in determining the virtual gene order within the partially sequenced genome. Genome zipper links the annotated and fully sequenced genome of sorghum, Brachypodium and rice with the data of less-studied species to predict the gene order and organization of the gene [113,114].
This entry is adapted from the peer-reviewed paper 10.3390/genes14061281
This entry is offline, you can click here to edit this entry!