Calcium signaling involves the movement of calcium ions within or between cells, which can affect the electrochemical gradients between intra- and extracellular membranes, ligand binding, enzyme activity, and other mechanisms that determine cell fate. Calcium signaling in muscle, as elucidated by the sliding filament model, plays a significant role in muscle contraction. However, as organisms age, alterations occur within muscle tissue. These changes include sarcopenia, loss of neuromuscular junctions, and changes in mineral concentration, all of which have implications for calcium’s role. Additionally, a field of study that has gained recent attention, cellular senescence, is associated with aging and disturbed calcium homeostasis, and is thought to affect sarcopenia progression.
- calcium
- aging muscle
- senescence
- aging
- calcium signaling
1. Introduction
2. Calcium Signaling and Aging Muscle
2.1. Role of Calcium in Muscle
2.2. Aging’s Effect on Muscle
| Author/Year | Species/Age | Finding | Implication | Ref. |
|---|---|---|---|---|
| Human Studies | ||||
| Goodpaster, B.H. et al. (2006) | Three-year changes in old (70–79 years; n = 1880) subjects (female, 51%; male, 48%) | Initially functioning older adults exhibited three-fold greater loss in strength than the loss of muscle mass over the course of 3 years. Maintenance or even gain of lean mass did not necessarily prevent loss of strength. |
Loss of strength is more rapid and suggests a decline in the quality of muscle. Losses of strength can increase risks of falls and serious injury. | [30] |
| Venturelli, M. et al. (2014) |
Young (25 ± 2 years; n = 12), old-mobile (87 ± 3 years of age; n = 12) and 12 old-immobile (88 ± 4 years; n = 12) sex-matched subjects (female, n = 9; male, n = 3) | Mean skeletal telomere length of thigh decreased with age, but not of arm. Mean free radical increased with age in thigh but not in arm. |
Chronological age does not affect the cellular aging of skeletal muscle evenly. Physical inactivity could be mediated by the free radical effect. | [27] |
| Murgia, M. et al. (2017) | Young (22–27 years; n = 4) and old (65–75 years of age, n = 4) non-sarcopenic subjects | Fiber size of fast-twitch (type 2a) but not slow-twitch (type 1) muscles decreased with age. Decreased respiratory chain complexes were found in aging muscles. Changes in protein quality, turnover, and metabolic pathways were changed with age in muscles. | Many glycolytic enzymes were expressed higher in slow fibers of the older cohort, and these same enzymes declined within fast-twitch fibers, showing changes in mitochondria in line with previous studies. These proteomic data support the idea that aging may differentially affect type 2 muscle fibers, protein homeostasis, mitochondria function, and metabolic pathways. | [31] |
| Walton, R.G. et al. (2019) | Randomized, double-blind trials in placebo (n = 55) and Metformin (n = 54) groups of old (over 65 years) subjects | Metformin inhibited progressive resistance training-induced lean mass gain but did not change the effect of weight loss from training. Metformin prevented decreases in type 1 fiber frequency. | Metformin inhibits gains in fat-free mass in response to concurrent aerobic and resistance training in subjects with prediabetes. Metformin may inhibit hypertrophy via mTORC1 inhibition. | [32] |
| Therakomen, V. et al. (2020) | Old (over 60 years, n = 330) male subjects | Development of sarcopenia is positively correlated with age, as is prefrailty and low physical activity. | The study supports previously understood risk factors for primary sarcopenia: age, prefrailty, physical activity, and nutritional status, but not sex. | [33] |
| Hester, G.M. et al. (2021) | Young (n = 15, age = 20.7 ± 2.2 years) and old (n = 15, age = 71.6 ± 3.9 years) male non-sarcopenic subjects | Peak torque was lower in the older group at all velocities compared to the young group. The whole-muscle cross-sectional area was smaller in old muscles. Type 1 fiber was larger and type 2 fiber was smaller in muscles in the older group. | Microbiopsy methods appear to be viable alternatives that are less intrusive but with similar results. The lack of association among neuromuscular junction deterioration, strength, and age-related muscle fiber atrophy may be due to the fact that samples were from a non-sarcopenic healthy elderly population. | [34] |
| Bres, E. et al. (2023) | Sarcopenic (n = 30) and non-sarcopenic (n = 22) old (over 70 years) subjects | Serum fibroblast growth factor (FGF) 19 was correlated with muscle ultrasound parameters of pennatation angle and muscle fiber length. FGF19 levels were not correlated with age, BMI, nutritional parameters, or tissue mass. | The association of FGF19 and the pennetation angle implies that a high-FGF19 environment promotes both the development of fast-twitch muscles as well as a negative association with balance and lower extremity strength, suggesting the role of FGF19 in muscle function and architecture. | [35] |
| Animal studies | ||||
| Lang, F. et al. (2018) | Manual denervation model of sarcopenia in adult C57BL/6J mice | Skeletal muscle exhibits varied protein changes after denervation, with opposing protein changes between type 1 and type 2a muscle fibers of Soleus during muscle atrophy. | Using a manual denervation method to study muscle atrophy, at 7 days post-denervation, this group showed complexities of response between different muscle fibers and tissues. | [36] |
| Lukjanenko, L. et al. (2020) | Young (9–13 weeks) and aged (20–25 months) C57BL/6J mice | Aging impaired fibro/adipogenic progenitor (FAP) functions with a failure to support muscle stem cells. Transcriptome analysis relieved the downregulated WNT1 inducible signaling pathway protein 1 (WISP1) gene comparing aged to young activated FAPs. |
Aging damages functions of FAPs and their ability to support myogenesis, the regenerative capacity. WISP1 is a FAP-derived factor controlling muscle stem cell expansion and differentiation, and age-induced loss of WISP occurs due to a lack of FAP population. | [37] |
| Xu, H. et al. (2021) | Whole body CuZnSOD KO mice and muscle-specific mitochondrial targeted catalase (mMCAT) transgenic mice with C57BL/6J background. accelerated sarcopenia model in female C57BL/6J mice (n = 12) |
An altered oxygen consumption rate and peroxide generation in CuZnSOD KO mice is reversed through mMCAT expression. Significant muscle loss and function is observed in CuZnSOD KO mice. Muscle fiber composition and diameter in CuZnSOD KO mice were decreased through mMCAT. |
In an accelerated sarcopenia model, mMCAT is sufficient to prevent the majority of muscle atrophy and weaknessin CuZnSOD KO mice. The absence of CuZnSOD leads to a reduction in force and disruption of neuromuscular junction with increased mitochondrial ROS. Mitochondrial scavenging capacity is important for the prevention of the loss of innervation, muscle atrophy, and weakness. |
[38] |
| Kim, K.H. et al. (2022) | Young (5 weeks) and aged (25 month) C57BL/6J female mice | Fecal microbiota transplantation of young mice improved grip strength, muscle fiber thickness, and other fitness markers. Young donor transplantation increased genes involved in cell differentiation, proliferation, and fatty acid synthesis in muscle. | A group of Bacteroidetes from the young-derived microbiota are discriminative in old mice, increasing gene expression in recipients’ muscles and skin. This supports that the age related changes in the microbiome can play a role with changes in aging muscles. | [39] |
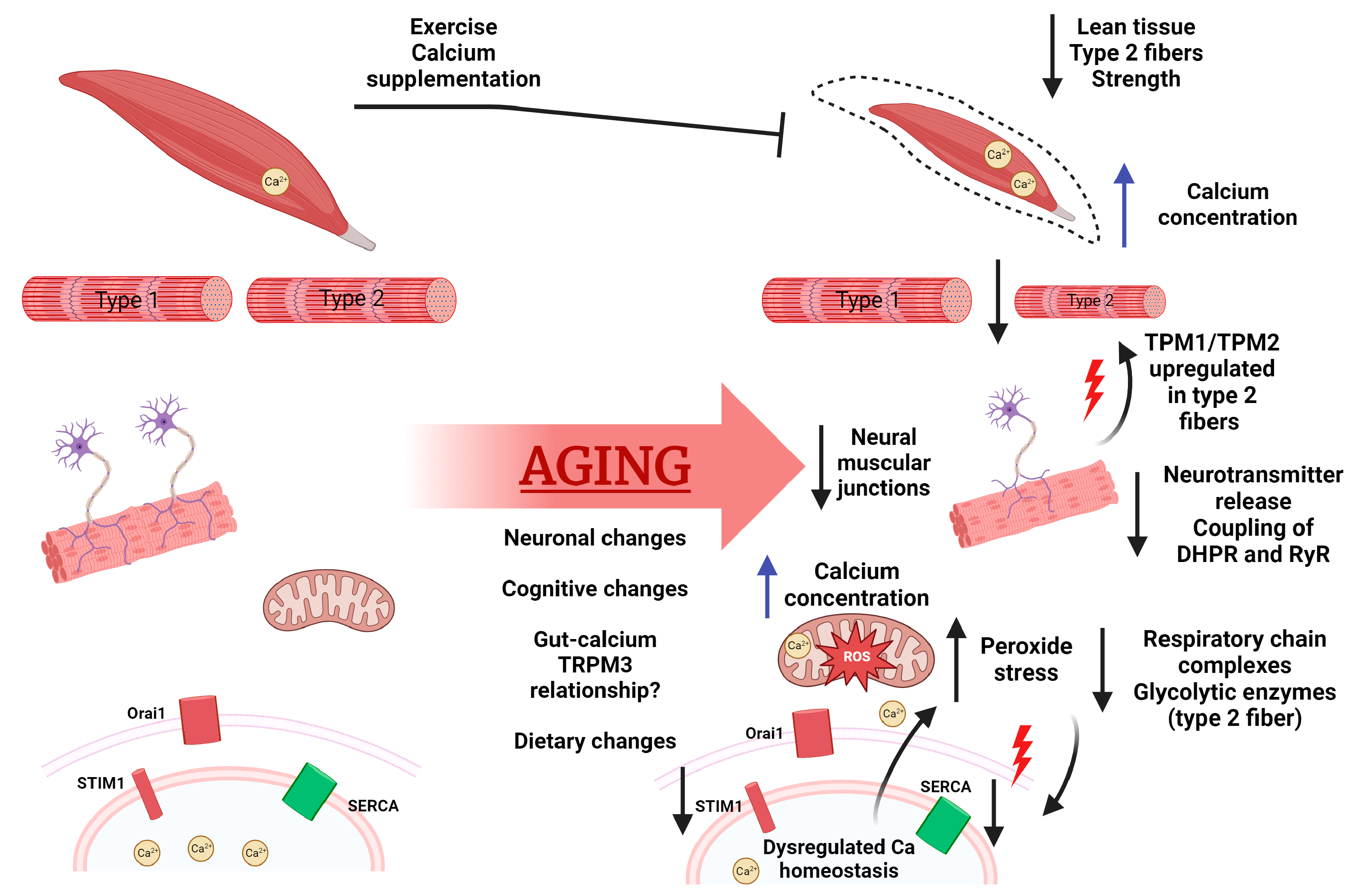
2.3. Aging’s Effect on Calcium
3. Cellular Senescence
3.1. Aging Muscle and Senescence
3.2. Mitochondria Dysfunction and Senescence
This entry is adapted from the peer-reviewed paper 10.3390/ijms242317034
References
- Arango-Lopera, V.E.; Arroyo, P.; Gutierrez-Robledo, L.M.; Perez-Zepeda, M.U.; Cesari, M. Mortality as An Adverse Outcome of Sarcopenia. J. Nutr. Health Aging 2013, 17, 259–262.
- Moiseeva, V.; Cisneros, A.; Sica, V.; Deryagin, O.; Lai, Y.; Jung, S.; Ortet, L.; Lukesova, V.; Volpe, G. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 2023, 613, 169.
- Zembron-Lacny, A.; Dziubek, W.; Wolny-Rokicka, E.; Dabrowska, G.; Wozniewski, M. The Relation of Inflammaging with Skeletal Muscle Properties in Elderly Men. Am. J. Mens. Health 2019, 13, 1557988319841934.
- Rosa, S.C.S.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607.
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Fraser, D.G.; Onken, J.L.; Johnson, K.O.; Verzosa, G.C.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246.
- Mijares, A.; Allen, P.D.; Lopez, J.R. Senescence Is Associated With Elevated Intracellular Resting in Mice Skeletal Muscle Fibers. An in vivo Study. Front. Physiol. 2021, 11, 601189.
- Martin, N.; Zhu, K.; Cxarnecka-Herok, J.; Vernier, M.; Bernard, D. Regulation and Role of Calcium in Cellular Senescence. Cell Calcium 2023, 110, 102701.
- Seturo, E.; Fumiko, E.; Ayako, K. Troponin as the Ca++-receptive protein in the contractile system. J. Biochem. 1967, 62, 137–138.
- Rossi, D.; Barone, V.; Giacomello, E.; Cusimano, V.; Sorrentino, V. Sarcoplasmic Reticulum: An Organized Patchwork of Specialized Domains. Traffic 2008, 9, 1044–1049.
- Chen, M.; Xu, D.; Wu, A.Z.; Kranias, E.; Lin, S.; Chen, P.; Chen, Z. Phospholamban regulates nuclear Ca2+ stores and inositol 1,4,5-trisphosphate mediated nuclear Ca2+ cycling in cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 123, 185–197.
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857.
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996.
- Foskett, J.K.; White, C.; Cheung, K.; Mak, D.D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 2007, 87, 593–658.
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57.
- Johnson, C.K.; Harms, G.S. Tracking and localization of calmodulin in live cells. Biochim. Biophys. Acta 2016, 1863, 2017–2026.
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121.
- Brunello, E.; Marcucci, L.; Irving, M.; Fusi, L. Activation of skeletal muscle is controlled by a dual-filament mechano-sensing mechanism. Proc. Natl. Acad. Sci. USA 2023, 120, e2302837120.
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153.
- Xu, C.; Craig, R.; Tobacman, L.; Horowitz, R.; Lehman, W. Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys. J. 1999, 77, 985–992.
- Pierantozzi, E.; Szentesi, P.; Paolini, C.; Dienes, B.; Fodor, J.; Oláh, T.; Colombini, B.; Rassier, D.E.; Rubino, E.M.; Lange, S.; et al. Impaired Intracellular Ca2+ Dynamics, M-Band and Sarcomere Fragility in Skeletal Muscles of Obscurin KO Mice. Int. J. Mol. Sci. 2022, 23, 1319.
- Randazzo, D.; Blaauw, B.; Paolini, C.; Pierantozzi, E.; Spinozzi, S.; Lange, S.; Chen, J.; Protasi, F.; Reggiani, C.; Sorrentino, V. Exercise-induced alterations and loss of sarcomeric M-line organization in the diaphragm muscle of obscurin knockout mice. Am. J. Physiol.-Cell Physiol. 2017, 312, C16.
- Bravo-Sagua, R.; Parra, V.; Muñoz-Cordova, F.; Sanchez-Aguilera, P.; Garrido, V.; Contreras-Ferrat, A.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum and calcium signaling in muscle cells: Homeostasis and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 197–264.
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970.
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97.
- Valdés, J.A.; Flores, S.; Fuentes, E.N.; Osorio-Fuentealba, C.; Jaimovich, E.; Molina, A. IGF-1 induces IP3-dependent calcium signal involved in the regulation of myostatin gene expression mediated by NFAT during myoblast differentiation. J. Cell. Physiol. 2013, 228, 1452–1463.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278.
- Venturelli, M.; Morgan, G.R.; Donato, A.J.; Reese, V.; Bottura, R.; Tarperi, C.; Milanese, C.; Schena, F.; Reggiani, C.; Naro, F.; et al. Cellular aging of skeletal muscle: Telomeric and free radical evidence that physical inactivity is responsible and not age. Clin. Sci. 2014, 127, 415–421.
- Bruce, A.; Alexander, J.; Julian, L.; Martin, R.; Keith, R. Walter, Peter, Genesis, Modulation, and Regeneration of Skeletal Muscle, Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2022.
- Coletta, G.; Phillips, S.M. An elusive consensus definition of sarcopenia impedes research and clinical treatment: A narrative review. Ageing Res. Rev. 2023, 86, 101883.
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylacsky, F.A.; Visser, M.; Newman, A.B. The Loss of Skeletal Muscle Strength, Mass, and Quality in Older Adults: The Health, Aging and Body Composition Study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064.
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409.
- Walton, R.G.; Dungan, C.M.; Long, D.E.; Tuggle, S.C.; Kosmac, K.; Peck, B.D.; Bush, H.M.; Villasante Tezanos, A.G.; McGwin, G.; Windham, S.T.; et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: A randomized, double-blind, placebo-controlled, multicenter trial: The MASTERS trial. Aging Cell 2019, 18, e13039.
- Therakomen, V.; Petchlorlian, A.; Lakananurak, N. Prevalence and risk factors of primary sarcopenia in community-dwelling outpatient elderly: A cross-sectional study. Sci. Rep. 2020, 10, 19551.
- Hester, G.M.; VanDusseldorp, T.A.; Ha, P.L.; Kiani, K.; Olmos, A.A.; Jabbari, M.; Kalladanthyil, S.; An, S.; Bailly, A.R.; Dalton, B.E.; et al. Microbiopsy Sampling for Examining Age-Related Differences in Skeletal Muscle Fiber Morphology and Composition. Front. Physiol. 2022, 12, 756626.
- Bres, E.; Bouvier, J.; Courtay, A.; Delaire, L.; Humblot, J.; Cuerq, C.; Tripoz-Dit-Masson, S.; Fauvernier, M.; Gilbert, T.; Bonnefoy, M. FGF19 and muscle architecture in older patients. Exp. Gerontol. 2023, 174, 112120.
- Lang, F.; Khaghani, S.; Turk, C.; Wiederstein, J.L.; Holper, S.; Piller, T.; Nogara, L.; Blaauw, B.; Gunther, S.; Muller, S.; et al. Single Muscle Fiber Proteomics Reveals Distinct Protein Changes in Slow and Fast Fibers during Muscle Atrophy. J. Proteome Res. 2018, 17, 3333–3347.
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Miglivacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433–446.e7.
- Xu, H.; Ranjit, R.; Richardson, A.; Van Remmen, H. Muscle mitochondrial catalase expression prevents neuromuscular junction disruption, atrophy, and weakness in a mouse model of accelerated sarcopenia. J. Cachexia Sarcopenia Muscle 2021, 12, 1582–1596.
- Kim, K.H.; Chung, Y.; Huh, J.; Park, D.J.; Cho, Y.; Oh, Y.; Jeong, H.; Yoon, J.; Kang, J.; Shin, H.; et al. Gut microbiota of the young ameliorates physical fitness of the aged in mice. Microbiome 2022, 10, 238.
- Shan, Z.; Rehm, C.; Rogers, G.; Ruan, M.; Wang, D.; Hu, F.; Mozaffarian, D.; Zhang, F.F. Bhupathiraju, Shilpa. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 2019, 322, 1178–1187.
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front. Aging Neurosci. 2014, 6, 208.
- Paintin, J.; Cooper, C.; Dennison, E. Osteosarcopenia. Br. J. Hosp. Med. 2018, 79, 253–258.
- Kemmler, W.; Kohl, M.; Fröhlich, M.; Jakob, F.; Engelke, K.; Stengel, S.; Schoene, D. Effects of High-Intensity Resistance Training on Osteopenia and Sarcopenia Parameters in Older Men with Osteosarcopenia—One-Year Results of the Randomized Controlled Franconian Osteopenia and Sarcopenia Trial (FrOST). J. Bone Min. Res. 2020, 35, 1634.
- Hill, T.R.; Verlaan, S.; Biesheuvel, E.; Eastell, R.; Bauer, J.M.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; Mets, T.; et al. A Vitamin D, Calcium and Leucine-Enriched Whey Protein Nutritional Supplement Improves Measures of Bone Health in Sarcopenic Non-Malnourished Older Adults: The PROVIDE Study. Calcif. Tissue Int. 2019, 105, 383–391.
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.; Sun, P.Y.; Davies, K.J.; Tilman, G. Sarcopenia—Molecular mechanisms and open questions. Ageing Res. Rev. 2021, 65, 101200.
- Dayal, A.; Schrötter, K.; Pan, Y.; Föhr, K.; Melzer, W.; Grabner, M. The Ca2+ influx through the mammalian skeletal muscle dihydropyridine receptor is irrelevant for muscle performance. Nat. Commun. 2017, 8, 1–14.
- Liu, Y.; Sugiura, Y.; Chen, F.; Lee, K.; Ye, Q.; Lin, W. Blocking skeletal muscle DHPRs/Ryr1 prevents neuromuscular synapse loss in mutant mice deficient in type III Neuregulin 1 (CRD-Nrg1). PLoS Genet. 2019, 15, e1007857.
- Edwards, J.N.; Blackmore, D.G.; Gilbert, D.F.; Murphy, R.M.; Launikonis, B.S. Store-operated calcium entry remains fully functional in aged mouse skeletal muscle despite a decline in STIM1 protein expression. Aging Cell 2011, 10, 675–685.
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle Aging Is Associated with Compromised Ca2+ Spark Signaling and Segregated Intracellular Ca2+ Release. J. Cell Biol. 2006, 174, 639–645.
- Grimes, D.; Johnson, R.; Pashos, M.; Cummings, C.; Kang, C.; Sampedro, G.R.; Tycksen, E.; McBride, H.J.; Sah, R.; Lowell, C.A.; et al. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 24403–24414.
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79.
- Yi, F.; Zhou, X.; Gumpper, K.; Zhu, H. MG29 Interacts with Bin-1 to Maintain T-Tubule Structure in Skeletal Muscle Physiology and Regeneration. FASEB J. 2019, 33, 868-24.
- Fraysse, B.; Desaphy, J.; Rolland, J.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Camerino, C.; Didonna, M.P.; Cocchi, D.; Luca, A.D.; et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol. Dis. 2006, 21, 372–380.
- Morel, J.; Sauzéat, L.; Goeminne, L.J.; Jha, P.; Williams, E.; Houtkooper, R.H.; Aebersold, R.; Auwerx, J.; Balter, V. The mouse metallomic landscape of aging and metabolism. Nat. Commun. 2022, 13, 607.
- Kim, Y.; Hong, K.; Han, K.; Park, Y.C.; Park, J.; Kim, K.; Kim, B.T. Longitudinal Observation of Muscle Mass over 10 Years According to Serum Calcium Levels and Calcium Intake among Korean Adults Aged 50 and Older: The Korean Genome and Epidemiology Study. Nutrients 2020, 12, 2856.
- Asghar, M.Y.; Törnquist, K. Transient Receptor Potential Canonical (TRPC) Channels as Modulators of Migration and Invasion. Int. J. Mol. Sci. 2020, 21, 1739.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Zhang, X.; Habiballa, L.; Aversa, Z.; Ng, Y.E.; Sakamoto, A.E.; Englund, D.A.; Pearsall, V.M.; White, T.A.; Robinson, M.M.; Rivas, D.A. Characterization of cellular senescence in aging skeletal muscle. Nat. Aging 2022, 2, 601.
- Tsuyoshi, K.; David, A.B.; Richard, W.; Tomas, P.A. Influences of aging and caloric restriction on the transcriptional profile of transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 5093–5098.
- Perez, K.; Ciotlos, S.; McGirr, J.; Limbad, C.; Doi, R.; Nederveen, J.P.; Nilsson, M.I.; Winer, D.A.; Evans, W.; Tarnopolsky, M. Single nuclei profiling identifies cell specific markers of skeletal muscle aging, frailty, and senescence. Aging 2022, 14, 9393–9422.
- Dungan, C.M.; Peck, B.D.; Walton, R.G.; Huang, Z.; Bamman, M.M.; Kern, P.A.; Peterson, C.A. In vivo analysis of γH2AX+ cells in skeletal muscle from aged and obese humans. FASEB J. 2020, 34, 7018.
- Demaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.; Laberge, R.M.; Vijg, J.; Steeg, H.V.; Dolle, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733.
- Prieto, L.; Graves, S.; Baker, D. Insights from In Vivo Studies of Cellular Senescence. Cells 2020, 9, 954.
- Raynard, C.; Tessier, N.; Huna, A.; Warnier, M.; Flaman, J.; Van Coppenolle, F.; Ducreux, S.; Martin, N.; Bernard, D. Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells. Int. J. Mol. Sci. 2022, 23, 9376.
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720.
- Zhou, J.; Dhakal, K.; Yi, J. Mitochondrial Ca2+ uptake in skeletal muscle health and disease. Sci. China Life Sci. 2016, 59, 770–776.
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A.M.K. Mitochondria in Health, Disease, and Aging. Physiol. Rev. 2023, 103, 2349–2422.
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, 1.
- Miwa, S.; Kashyap, S.; Chini, E.; Von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447.
- Li, Y.; Tran, Q.; Shrestha, R.; Piao, L.; Park, S.; Park, J.; Park, J. LETM1 is required for mitochondrial homeostasis and cellular viability. Mol. Med. Rep. 2019, 19, 3367–3375.
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156.
- Harrington, J.L.; Murphy, E. The mitochondrial calcium uniporter: Mice can live and die without it. J. Mol. Cell. Cardiol. 2015, 78, 46–53.
- Young, M.P.; Schug, Z.T.; Booth, D.M.; Yule, D.I.; Mikoshiba, K.; Hajnoczky, G.; Joseph, S.K. Metabolic adaptation to the chronic loss of Ca2+ signaling induced by KO of IP3 receptors or the mitochondrial Ca2+ uniporter. J. Biol. Chem. 2022, 298, 101436.
- Sena, L.; Chandel, N. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167.
- Yamamoto-Imoto, H.; Minami, S.; Shioda, T.; Yamashita, Y.; Sakai, S.; Maeda, S.; Yamamoto, T.; Oki, S.; Takashima, M.; Yamamuro, T.; et al. Age-associated decline of MondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Rep. 2022, 38, 110444.
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnoczky, G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019, 29, 1274–1286.e6.
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193.