Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Cisplatin is a commonly used chemotherapeutic agent with proven efficacy in treating various malignancies, including testicular, ovarian, cervical, breast, bladder, head and neck, and lung cancer. Cisplatin is also used to treat tumors in children, such as neuroblastoma, osteosarcoma, and hepatoblastoma.
- cisplatin
- ototoxicity
- hearing loss
- cochlea
1. Introduction
Platinum-based chemotherapeutic drugs, such as cisplatin, carboplatin, and oxaliplatin, are widely used in the treatment of solid malignant tumors in both adult and pediatric patients, including testicular, ovarian, cervical, breast, bladder, head and neck, and lung cancer. Cisplatin (cis-diamminedichloroplatinum (II)) is the most widely used platinum-based drug. Since its approval by the U.S. Food and Drug Administration (FDA) in 1978, cisplatin has been a core therapy in oncology and an essential systemic therapy for germ cell malignancies [1].
Cisplatin induces the crosslinking of DNA and the formation of DNA adducts, which ultimately trigger apoptosis in cancer cells [2,3]. Despite its clinical effectiveness, cisplatin can cause severe side effects due to its non-specific mechanisms of action, targeting both cancer cells and healthy tissues. The adverse effects of cisplatin are dose-dependent and include ototoxicity, nephrotoxicity, neurotoxicity, hepatotoxicity, gastrointestinal toxicity, and retinal toxicity. Ototoxicity is the most common side effect due to the accumulation and prolonged retention of cisplatin within cochlear tissues [4].
Cisplatin ototoxicity mainly arises from effects on the sensory hair cells, spiral ganglion neurons (SGNs), and secretory and connective tissues (stria vascularis and spiral ligament) of the cochlea (Figure 1) [2]. Due to the absence of regenerative capacity in mammalian hair cells and SGNs, their death is irreversible, resulting in permanent sensorineural hearing loss. Cisplatin-induced ototoxicity is characterized by bilateral, moderate-to-profound high-frequency hearing loss, and a significant loss of outer hair cells (OHCs) in the basal turn of the cochlea. Although cisplatin primarily affects auditory function, inner ear toxicity can also present as tinnitus and infrequently as ear pain or balance disorder due to vestibulotoxicity [5,6]. Ototoxicity is determined by several factors, including the patient’s age, cumulative cisplatin dose, and predisposition [7]. Cisplatin-induced hearing loss affects 40–60% of adults, of which 18% have severe to profound hearing loss after cisplatin treatment, whilst tinnitus is present in 40% of cases [8]. Up to 60% of children treated with cisplatin are also affected by hearing loss [9]. Pediatric patients may experience speech and language development difficulties due to hearing impairments. Other consequences of cisplatin-induced hearing loss include social isolation, anxiety, and depression, negatively affecting overall well-being.
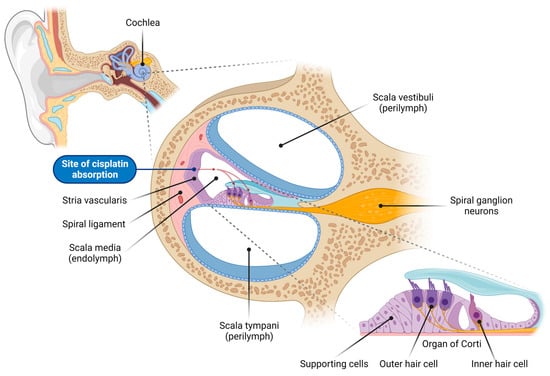
Figure 1. Overview of the cochlear structure and the site of cisplatin absorption. This illustration depicts the anatomical structure of the cochlea, the organ of hearing located within the inner ear, highlighting the various cell types vulnerable to cisplatin-induced damage. The cochlea consists of three fluid-filled compartments: scala vestibuli and scala tympani, filled with Na+-rich perilymph, and scala media, filled with K+-rich endolymph. The scala media houses the organ of Corti, which comprises the inner hair cells (responsible for auditory transduction), outer hair cells (critical for cochlear amplification), and surrounding supporting cells. Adjacent to the organ of Corti sits the lateral wall, comprising the stria vascularis (responsible for generating and maintaining the endocochlear potential, the driving force for sensory transduction) and spiral ligament (supporting the stria vascularis and cochlear fluid homeostasis). Spiral ganglion neurons innervate the sensory hair cells and transmit auditory signals to the auditory nuclei in the brainstem. Cisplatin enters the endolymph in the scala media through capillaries in the stria vascularis, and is subsequently absorbed by the sensory hair cells in the organ of Corti (red dashed arrows).
In addition to lowering the cisplatin dose or transitioning to a non-cisplatin treatment, both of which reduce the efficacy of chemotherapy, interventions that can prevent or restore hearing loss induced by cisplatin chemotherapy are lacking. Currently, managing cisplatin-induced hearing loss mainly involves the use of hearing aids. Recently, the antioxidant sodium thiosulfate (Pedmark) was approved by the U.S. FDA to prevent cisplatin-induced hearing loss in pediatric patients; however, its efficacy remains limited as 28–33% of the patients still suffered hearing loss [10,11,12,13]. Thus, there is still an unmet clinical need for interventions to manage cisplatin ototoxicity. Several promising preventative and therapeutic approaches are currently in various stages of preclinical or clinical development.
2. Oxidative Stress in Cisplatin-Induced Ototoxicity
Oxidative stress is a key contributor to the complex pathways responsible for cisplatin-induced cochlear injury and hearing loss. Extensive evidence implicates the increased production of reactive oxygen species (ROS), which triggers mitochondrial-mediated apoptosis in cochlear hair cells [7,19,21]. ROS are derivatives of molecular oxygen that occur as natural by-products of oxidative phosphorylation in mitochondria. Although ROS play a crucial role in cellular signaling in a variety of physiological processes, an intricate system of endogenous antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH), regulates ROS production and metabolism. An imbalance between ROS production and the level of antioxidants leads to oxidative stress, which can cause irreversible ROS-mediated damage to cellular DNA, proteins, and lipids [33]. In cochlear tissues with high metabolic demands, ROS-induced oxidative stress impacts mitochondrial DNA (mtDNA), respiratory chain proteins, and mitochondrial membranes, resulting in mtDNA mutations, protein oxidation, and lipid peroxidation, respectively [33,34,35,36,37]. This damage leads to mitochondrial dysfunction, which can facilitate further ROS production in a positive feedback loop.
Cisplatin increases the production of ROS in the cochlea primarily by activating NADPH oxidase-3 isoform (NOX3), a superoxide-generating enzyme highly expressed in the inner ear [38,39,40]. Cisplatin increases the expression of NOX3 in the supporting cells and OHCs, particularly in the basal turn of the cochlea [39]. The upregulation of NOX3 correlates with cisplatin-induced cochlear damage and hearing loss [39]. In contrast, suppression of NOX3 by gene knockout or short interfering RNA (siRNA) protects against cisplatin-induced ototoxicity [39,41].
Another superoxide-producing enzyme, xanthine oxidase (XO), has also been implicated as a source of cisplatin-induced ROS generation in the cochlea [38,42]. XO and its natural substrate, hypoxanthine, increase the intracellular calcium concentration in OHCs, which can modulate OHC electromotility [43]. Both ROS-producing enzymes (NOX3 and XO) set off a cascade of events involving apoptotic signaling pathways, ultimately leading to apoptosis and functional loss in the cochlea.
Cisplatin’s impact on antioxidant enzyme levels further exacerbates its ototoxic effects, as it directly reduces the levels of antioxidants, such as SOD, CAT, glutathione reductase (GR), and glutathione peroxidase (GSH-Px) in the cochlea [44,45]. ROS overproduction and the depletion of endogenous antioxidants in the cochlea ultimately lead to lipid peroxidation, as evidenced by elevated levels of malondialdehyde and 4-hydroxynonenal [44,45,46].
Cisplatin also contributes to increased nitric oxide (NO) production in the cochlea by upregulating inducible nitric oxide synthase (iNOS) levels. NO causes the nitration of cochlear proteins, severely disrupting their normal function [47,48,49]. NO reacts with NOX3-generated superoxide to form the highly reactive oxidant peroxynitrite, which reacts with proteins to form nitrotyrosine, a common marker of oxidative damage [48]. Cochlear protein nitration correlates with the dose-dependent increase in cisplatin-induced cochlear damage, indicating the pivotal role of protein nitration in cisplatin ototoxicity and hearing loss [48]. Cisplatin-induced nitrative stress in the cochlea primarily induces the nitration of a specific protein, LIM Domain Only 4 (LMO4), a transcriptional regulator that controls the choice between cell survival and death in OHCs, SGNs, and strial cells [48,50]. Nitration reduces the cochlear expression of LMO4, ultimately contributing to cisplatin-induced cochlear apoptosis and ototoxicity [48]. This was corroborated in LMO4 conditional knockout mice, which displayed enhanced susceptibility to cisplatin-induced apoptosis and hearing loss [50].
Recent evidence suggests that ferroptosis, a newly discovered non-apoptotic form of programmed cell death characterized by an iron-dependent accumulation of lipid peroxides and reduced mitochondrial membrane potential, also plays a role in cisplatin-induced ototoxicity [51,52]. The inhibition of ferroptosis with ferrostatin-1, a ferroptosis inhibitor, was shown to markedly attenuate cisplatin-induced hair cell damage by inactivating lipid peroxide radicals and preserving mitochondrial function [51,52].
Other evidence suggests that cisplatin causes a dysregulation of mitochondrial calcium homeostasis [53]. A recent study demonstrated that cisplatin acutely disrupts mitochondrial bioenergetics within the hair cells of the zebrafish lateral line by increasing mitochondrial activity and calcium levels [54]. The zebrafish lateral line is a valuable model for studying the roles of mitochondria in sensory hair cell pathologies and developing therapeutic strategies to prevent sensorineural hearing loss in humans. These alterations in mitochondrial function are also associated with elevated ROS levels and the activation of caspase-3-mediated apoptosis, indicating that mitochondrial dysfunction is an early event in the development of cisplatin-induced ototoxicity.
3. Antioxidant and Anti-Inflammatory Treatments for Cisplatin-Induced Ototoxicity
Several potential therapies have been explored in preclinical (animal models and cell lines) and clinical studies to prevent or attenuate cisplatin-induced ototoxicity by targeting oxidative stress and cochlear inflammation [101,102,103,104,105]. These approaches hold promise for the development of treatments to alleviate the ototoxic effects of cisplatin and prevent hearing loss. The main condition is that any treatment that prevents cisplatin ototoxicity should not interfere with its tumor-killing activity. Local intratympanic otoprotectant delivery has become an increasingly attractive proposition to prevent cisplatin-induced hearing loss compared with systemic administration. Local delivery bypasses the BLB and achieves higher drug concentrations in the cochlea without systemic side effects. Other innovations in drug delivery systems include nanoparticles, hydrogels, and environmental stimuli systems applied to the inner ear [7].
The antioxidant sodium thiosulfate (Pedmark, Fennec Pharmaceutical Inc., Research Triangle Park, NC, USA) recently became the first FDA-approved treatment for preventing cisplatin-induced hearing loss in pediatric patients aged 1 month and older with localized, non-metastatic solid tumors [10,13]. This approval followed two Phase 3 clinical trials (SIOPEL 6, ClinicalTrial.gov identifier: NCT00652132 and COG ACCL0431, ClinicalTrials.gov identifier: NCT00716976) where sodium thiosulfate (STS) was administered intravenously over 15 min, starting 6 h after cisplatin chemotherapy [11,12,106,107]. STS significantly reduced the incidence of hearing loss in children aged 1 month to 18 years by acting through two distinct mechanisms. Firstly, STS acts as an ROS scavenger and increases the levels of endogenous antioxidants after entering hair cells via sodium sulfate cotransporter 2 [13,108]. Secondly, it directly interacts with cisplatin, neutralizing its ototoxic effects [13,108]. The delayed administration of STS after cisplatin treatment enables cisplatin to exert its anti-cancer properties before being neutralized by STS [13,108,109].
N-acetyl cysteine (NAC) functions as both a direct free radical scavenger and a substrate for the synthesis of the antioxidant glutathione. Although it has demonstrated efficacy in preventing cisplatin-induced ototoxicity in animals when administered intratympanically [110], human studies have yielded mixed results with intratympanic NAC administration [111,112]. The optimal dosage and efficacy of intratympanic NAC injection in reducing cisplatin-induced hearing loss in head and neck cancer patients is being evaluated in a current Phase 2 clinical trial (ClinicalTrials.gov identifier: NCT04291209) [113]. Interestingly, a recent study [114] has suggested that mannitol, a diuretic that transiently increases the BLB permeability, can enhance the otoprotective effects of NAC and STS.
Ebselen, a synthetic mimic of the antioxidant enzyme GPx1 with anti-inflammatory properties, has also demonstrated beneficial effects against cisplatin ototoxicity [42,45,115,116,117]. Currently, SPI-1005, a proprietary oral formulation of ebselen, is undergoing Phase 2 clinical trials (ClinicalTrials.gov Identifier: NCT01451853) [118] to assess its potential in preventing and treating cisplatin-induced hearing loss and tinnitus. This clinical trial is aiming to evaluate the safety and efficacy of three different doses of SPI-1005 administered orally to patients diagnosed with head and neck or non-small cell lung cancer.
Another antioxidant that has shown promising results is D-methionine. This sulfur-containing amino acid acts as a direct scavenger of free radicals and protects the enzymatic activity of endogenous antioxidants. In animal studies, D-methionine demonstrated protective effects against cisplatin-induced ototoxicity through systemic [44,119,120,121] and local administration onto the round window membrane [122,123]. Campbell et al. [119] established that both oral and injected D-methionine yielded comparable levels of otoprotection, effectively preventing cisplatin-induced auditory brainstem response (ABR) threshold shifts. Similarly, a Phase 2 clinical trial involving cancer patients revealed that those who received oral D-methionine prior to each cisplatin dose showed reduced ABR threshold shifts compared with the placebo group [124].
Dexamethasone, a corticosteroid with anti-inflammatory properties, has shown some degree of protection against cisplatin ototoxicity in animal studies after intratympanic administration [125,126,127,128,129,130,131,132,133]. However, clinical studies have demonstrated limited otoprotective effects of intratympanic dexamethasone [134,135]. The most recent clinical trial (ClinicalTrials.gov identifier: NCT02997189) [136] investigating OTO-104, a sustained-release hydrogel formulation of dexamethasone, in preventing cisplatin-induced ototoxicity was terminated due to negative efficacy outcomes in a related clinical study.
Due to the pivotal role of the pro-inflammatory cytokine TNF-α in the development of cisplatin-induced ototoxicity, preclinical studies have demonstrated the otoprotective effect of the TNF-α inhibitor etanercept. Rats that received intratympanic etanercept prior to cisplatin administration exhibited significantly reduced ABR threshold shifts and increased OHC survival compared with non-treated controls [96]. Interestingly, etanercept injection also led to significant decreases in both serum and cochlear mRNA and protein levels of not only TNF-α, but also IL-1β and IL-6, suggesting that TNF-α contributes to the expression of other pro-inflammatory cytokines [81]. These findings demonstrate the potential of TNF-α inhibition as a therapeutic approach to preventing cisplatin-induced hearing loss [96].
Controlling inflammation by regulating STAT1 and STAT3-dependent pathways in the cochlea could serve as an effective therapeutic approach for cisplatin-induced ototoxicity, as evidenced by various preclinical studies [93,94,96,137,138]. The inhibition of STAT1 using short interfering RNA (siRNA) reduced the expression of pro-inflammatory mediators and cell apoptosis in the rat cochlea, protecting against ototoxicity [96]. Intratympanic administration of R-phenylisopropyladenosine R-PIA, an adenosine A1 receptor agonist, protected against OHC damage and hearing loss by reducing ROS production and STAT1-mediated inflammation [137,139]. Cisplatin ototoxicity in rats was also reduced by adenosine amine congener (ADAC), acting on adenosine A1 receptors [140]. The phytopharmaceutical aucubin, a member of the iridoid glycoside family that exhibits antioxidant and anti-inflammatory properties, protects against cisplatin-induced cochlear damage in vitro and in vivo by activating the STAT3 pathway [138]. Direct activation of the transient receptor potential vanilloid 1 (TRPV1) channel with capsaicin, a TRPV1 agonist extracted from chili peppers, increased the ratio of phosphorylation-activated STAT3/STAT1, leading to an anti-inflammatory response and protection from cisplatin ototoxicity [93]. Furthermore, apelin-13, a peptide hormone and endogenous ligand of the apelin receptor APJ, protected against cisplatin-induced ototoxicity and inhibited ROS production, apoptosis, and pro-inflammatory cytokine expression by reducing the phosphorylation and activation of STAT1 while increasing the phosphorylation and activation of STAT3 [94].
Other studies suggest that some polyphenols can modulate both oxidative stress and inflammation in the cochlea [141]. The polyphenol curcumin reduced cisplatin-induced hearing loss and suppressed NF-κB-related inflammation pathways in the cochlea while providing optimal chemosensitivity [141,142]. Recently, treatment with avenanthramide-C (AVN-C), a potent naturally occurring polyphenolic compound found exclusively in oats, reduced ROS production, enhanced the survival of OHCs and inner hair cell (IHC) presynaptic ribbons, and protected against cisplatin-induced hearing loss in mice [143]. When administered to a mouse auditory HEI-OC1 cell line prior to cisplatin exposure, AVN-C significantly reduced ROS production and mitigated cisplatin-induced inflammation by reducing the expression of the pro-inflammatory mediators, IL-6, IL-1β, TNF-α, iNOS, and COX2 [143]. Observations from previous studies suggest that AVN-C decreases the secretion of pro-inflammatory cytokines by inhibiting both NF-κB activity and mast cell degranulation [144,145]. These findings suggest AVN-C is a potential therapeutic candidate for ameliorating cisplatin-induced oxidative stress and cochlear inflammation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242216545
This entry is offline, you can click here to edit this entry!