Human cytomegalovirus (HCMV) expresses a variety of viral regulatory proteins that undergo close interaction with host factors including viral-cellular multiprotein complexes. The HCMV protein kinase pUL97 represents a viral CDK ortholog (vCDK) that determines the efficiency of HCMV replication via phosphorylation of viral and cellular substrates. A hierarchy of functional importance of individual pUL97-mediated phosphorylation events has been discussed, however, the most pronounced pUL97-dependent phenotype could be assigned to viral nuclear egress, as illustrated by genetic ORF-UL97 deletion or pharmacological pUL97 inhibition. Despite earlier data pointing to a cyclin-independent functionality, experimental evidence increasingly emphasized the role of pUL97-cyclin complexes. Consequently, the knowledge about pUL97 involvement in host interaction, viral nuclear egress and additional replicative steps led to the postulation of pUL97 as an antiviral target. Indeed, validation experiments in vitro and in vivo confirmed the sustainability of this approach. Consequently, current investigations of pUL97 in antiviral treatment go beyond the known pUL97-mediated ganciclovir prodrug activation and henceforward include pUL97-specific kinase inhibitors. Among a number of interesting small molecules analyzed on experimental and preclinical stages, maribavir is presently investigated in clinical studies and, in the near future, might represent a first kinase inhibitor applied in the field of antiviral therapy.
- human cytomegalovirus (HCMV)
- protein kinase pUL97
- kinase-host interactions
- cyclin-dependent kinase (CDK) complexes
- regulatory mechanisms
- antiviral drugs
The Present Status of Controlling HCMV as a Major Human Pathogen
Molecular Biology of HCMV and Its Lytic Replication in Permissive Cells
Pathogenesis of HCMV Infection
Current Options of Prevention and Control
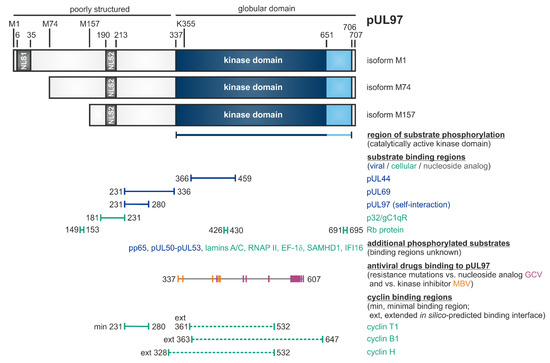
HCMV-Encoded Protein Kinase pUL97, a Multifunctional CDK Ortholog (vCDK)
Characteristics of the HCMV-Encoded Protein Kinase
| Property | General Description | Specific Feature | Own Findings (MM lab.) | Various References |
|---|---|---|---|---|
| Type of kinase | Ser/Thr | target site P + 5, target site LxSP |
[51,53,56,62,63,64] | [65,66,67,68,69] |
| Molecular mass, basic features | 100/80/70 kDa | isoforms due to alternative translational start sites | [44,45,54] | [43,52,67] |
| Expression pattern | three isoforms M1, M74, M15 (referring to other herpesviral protein isoforms) | differences in substrate binding, nuclear translocation and drug susceptibility | [44,70] | [71,72,73,74] |
| Similarity and sequence conservation with other kinases | low | <35% identity with herpesviral kinases, <15% identity with cellular kinases | [45,63] | [48,49,50,75] |
| Sequence conservation ORF-UL97 of HCMVs | high | no variation of translational start sites, NLS sequences or kinase domains | [44] | [76,77] |
| Related to cell kinases | cyclin-dependent kinases (CDKs), viral CDK ortholog | functional overlap with CDKs, specific crosstalk with CDK9, CDK7 and CDK1, direct interaction with cyclins | [47,55,56,78,79,80,81,82,83] | [57,84,85] |
| Coregulation of viral replication by pUL97 and cellular kinases | several novel cellular kinases, including CDKs, identified to be involved in HCMV replication | virus-supporting functions in signaling pathways and nuclear capsid egress | [55,56,86,87] | [88,89,90,91,92,93] |
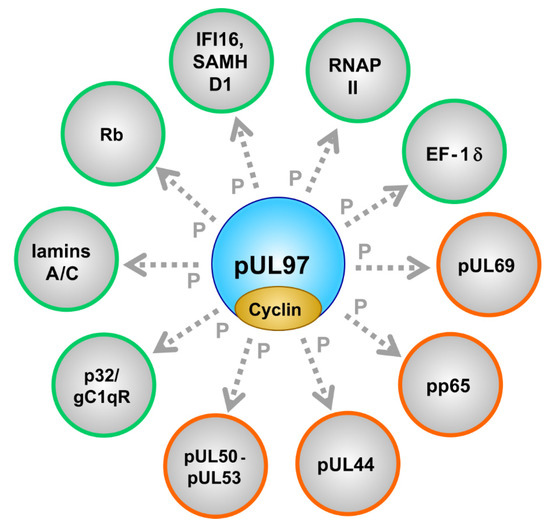
| Substrate proteins | viral, cellular | pUL44, pUL69, pp65, Rb, p32/gC1qR, nuclear lamins, EF-1δ, RNAP II, IFI16, SAMHD1 | [53,79,87,94,95,96,97,98] (references therein) | [57,75,84,99,100,101,102,103,104,105,106] (see also refs. in Figure 1) |
| Involvement in intrinsic immunity evasion | stimulation of viral counterdefense of immunity | interaction with cellular restriction factors IFI16 and SAMHD1 | [96,107] | [108] |
| Auto-phosphorylation | pronounced auto-phosphorylation activity, several N-terminal Ser and Thr residues | autophosphorylation most probably required for kinase activity/autoactivation | [44,54,56,94,109] | [65,66,110] |
| Nucleoside phosphorylation | ganciclovir, valganciclovir, penciclovir, acyclovir, etc. | prodrug-activating monophosphorylation as an essential step in antiviral therapy | [51,111] | [59,112,113,114,115] |
| Incorporation into virions | component of virion tegument | virion-derived pUL97 possesses highly detectable kinase activity | [45,95] | [43,116,117] |
| Intracellular localization | mainly nuclear | two nuclear localization signals, NLS-1 (6–35), NLS-2 (164–213), classical importin-α pathway | [45,46,63,97,118] | [60] |
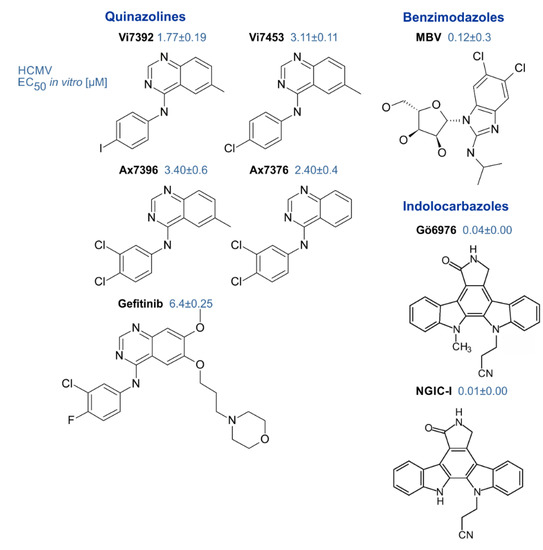
| Inhibitors of pUL97 | small molecules (<500 Da, various chemical classes) | indolocarbazoles, benzimidazoles, quinazolines, others | [53] (references therein) [44,64,119] | [114,120,121] |
| Phenotype of pUL97 inhibition or UL97 deletion | strongly reduced viral replication efficiency (100–1000-fold) | delayed replication kinetics; impaired genomic replication; impaired viral nuclear egress | [44,51,53,94,109,122,123] | [59,61,104,124] |
| A HCMV-Infected Cells | B Recombinant Expression | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cyclin Types | Cyclin IP MS|Wb | pUL97 IP MS|Wb | Colocalization in IF | Transfection | Yeast Two-Hybrid System | Phosphorylation by pUL97 in IVKA | |||
| B-like | Cyclin A | + | ± | - | - | . | . | . | . |
| Cyclin B1 | + | + | + | - | + | + | . | + | |
| Cyclin B2 | - | + | - | - | - | . | . | . | |
| Cyclin D1 | - | - | - | - | - | - | . | . | |
| Cyclin E | ± | ± | - | - | . | . | . | . | |
| Cyclin F | . | ± | - | - | . | . | . | . | |
| C-like | Cyclin H | + | + | - | - | + | - | - | - |
| Cyclin K | . | + | - | - | . | . | . | . | |
| Cyclin L2a | . | - | - | - | . | . | . | . | |
| Cyclin T1 | + | + | + | + | + | + | + | - | |
| Y-like | Cyclin Y | . | ± | - | - | . | . | . | . |
*Data on pUL97-cyclin interaction were derived from the experimental settings of either mass 188 spectrometry-based proteomics (MS) or Western blot detection (Wb), both performed by the use of 189 coimmunoprecipitates derived from cyclin-specific immunoprecipitation (cyclin IP) or pUL97 190 immunoprecipitation (pUL97 IP). Colocalization patterns between pUL97 and individual cyclins, in 191 particular nuclear punctate patterns of accumulation in viral replication centers, were determined by 192 indirect immunofluorescence (IF) double-stainings and confocal imaging. Recombinant expression of 193 pUL97 and/or cyclins was performed by transient transfected of 293T cells (transfection), yeast cells 194 (yeast two-hybrid assay) or bacterial expression systems, the latter for analyzing the phosphorylation 195 of recombinant cyclins by transfection-derived pUL97 in the in vitro kinase assay (IVKA). In panel A, 196 the criteria of categorization were set as follows: +, strong pUL97-cyclin interaction (MS: WSC ≥4; Wb: 197 % IP values > 20% IP control and ≥15-fold above Flag neg. control); ±, weak interaction (MS: WSC = 3; 198 Wb: % IP values >20% IP control or ≥15-fold above Flag neg. control); -, no detectable interaction; ., 199 not determined.
Phosphorylation of a Panel of Regulatory Viral Proteins and Host Factors through pUL97
| Protein Origin | Designation | Function | Remarks | References |
|---|---|---|---|---|
| Viral | pUL50 | core nuclear egress protein (NEC) | forms the NEC groove, multiple PPIs, phosphorylated by viral and cellular kinases | [62,99,126,127,134,135] |
| Viral | pUL53 | core nuclear egress protein (NEC) | forms NEC hook, possibly docking to capsids, phosphorylated by viral kinase | [99,136,137] |
| Viral | pUL44 | DNA polymerase pUL54 processivity factor | phosphorylation might regulate activity | [104,122] |
| Viral | pp65 | major tegument protein | massively phosphorylated and virion-associated with pUL97 | [44,60,95] |
| Viral | pUL69 | RNA transport regulator | phosphorylation regulates activity | [78,79,138,139] |
| Viral | pUL97 | CDK-like serine/threonine protein kinase, multifunctional | dimers/oligomers, autophosphorylation | [50,53,54,65,110,114,132] |
| Cellular | p32/gC1qR | multiligand binding protein, multifunctional | NEC bridging factor | [94,98,140] |
| Cellular | lamins A/C | structural and regulatory components of the nuclear envelope | lamin phosphorylation is a rate-limiting step of viral nuclear egress | [57,84,94,97,129,141] |
| Cellular | Rb | retinoblastoma protein, cell cycle check-point regulator | multiply phosphorylated by CDKs and pUL97 | [48,57,85,103,106] |
| Cellular | IFI16 and SAMHD1 | intrinsic immune restriction factors of virus infections | interferon-induced, phosphorylation-controlled | [96,105,107,108,142] |
| Cellular | RNAP II | main cellular mRNA transcriptase | activity-regulated by C-terminal phosphorylation (CTD) | [59,61,90,100] |
| Cellular | EF-1 | translation elongation factor 1 delta | activity-regulated by phosphorylation | [53,101] |
| Cellular | cyclins | regulatory subunits of CDKs | types B1, H, T1 were found pUL97-associated (possibly also B2, K, others) | [55,56,58,84,92] |
HCMV pUL97 and Related Herpesviral vCDKs
| Kinase Characteristics | pUL97 | CDK1 | CDK7 | CDK9 |
|---|---|---|---|---|
| Amino acids (aa) | 707 | 297 | 345 | 372 |
| Aa sequence identity to pUL97 | 100% | 4.5% | 4.2% | 8.6% |
| Cyclin binding partner [56,146,147] | cyclin B1 cyclin H cyclin T1 |
cyclin A1/A2 cyclin B1/B2/B3 cyclin D1/D3 cyclin F cyclin K (activating) |
cyclin H cyclin A2 cyclin B1/B2 cyclin E (activating) |
cyclin T1/T2 cyclin H cyclin K (activating) |
| Region in the kinase required for cyclin binding [55,148,149] | cyclin T1: 231ESQDSAVASGPGRIPQPLSGSSGEESATAVEADSTSHDDVHCTCSNDQII280 and in silico-predicted binding interfaces for cyclins B1, H and T1 spanning aa 328–647 | cyclin B1: a positively charged region in the N-lobe (containing K6, K9, K34, R36, R75, excluding the PSTAIRE helix) cyclin A2: 45PSTAIRE51 |
cyclin H: 56NRTALRE62 | cyclin T1/T2, K: 60PITALRE66 |
| Cyclin phosphorylation [56,82,146,150,151,152,153] | cyclin B1 | cyclin B1 S126 by CDK1 S128 by CDK1 | cyclin H by CDK7/CDK8-cyclin C (inhibitory) | n.d.* |
| T-loop phosphorylation [56,154,155,156,157,158,159,160,161] | no, (possibly S483) | T161 by CAK (activating) | S164 and T170 by CDK1/CDK2 (activating) | T186 by CaMK1D or CDK9 (S175 by CAK, not essential for activity) |
| Autophosphorylation [110,155,156] | yes | no | (yes) outside the T-loop | yes within the T-loop |
| Rb phosphorylation [66,75,82,145,162,163] | S780, S807, S811, T821, T823, T826 | S249, T252, T373, S807, S811 | no | C-terminus (793–834) |
| p53 phosphorylation [164,165,166] | n.d. | S315 | S33 (MAT1-dependent) | S33, S315, S392 |
| Lamin A/C phosphorylation [84,141,167,168] | S22 (inhibitory) | S22, S392 (inhibitory) | no | no |
| CTD RNAP II phosphorylation [100,169,170] | S2, S5 (activating) | no | S2, S5, S7 (activating) | S2, S5, S7 (activating) |
| SAMHD1 phosphorylation [171,172,173] | yes | T592 | n.d. | n.d. |
| HCMV pUL69 phosphorylation [78,79] | yes | yes | yes | yes |
| HCMV pUL50 phosphorylation [127] | yes | yes | n.d. | n.d. |
Validation of vCDK pUL97 as an Antiviral Target and Various pUL97 Inhibitors Explored as Experimental Antiviral Drugs
Role of the pUL97 Kinase in Anti-HCMV Standard Therapy
Target Validation and pUL97 Inhibitors
Clinical Investigation of the First Prototype of a Kinase Inhibitor in Antiviral Treatment: Maribavir
The Relevance of Targeting a Herpesviral Kinase Activity in Antiviral Strategies
- Lachmann, R.; Loenenbach, A.; Waterboer, T.; Brenner, N.; Pawlita, M.; Michel, A.; Thamm, M.; Poethko-Muller, C.; Wichmann, O.; Wiese-Posselt, M. Cytomegalovirus (CMV) seroprevalence in the adult population of Germany. PLoS ONE 2018, 13, e0200267. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1960–2014. [Google Scholar]
- Roizman, B.; Knipe, D.M. Herpesviruses and Their Replication; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 2399–2460. [Google Scholar]
- Stinski, M.F. Molecular Biology of Cytomegaloviruses. In The Herpesviruses; Viruses, B.R., Ed.; Springer: Boston, MA, USA, 1983; pp. 67–113. [Google Scholar]
- Lee, C.P.; Chen, M.R. Escape of herpesviruses from the nucleus. Rev. Med. Virol. 2010, 20, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef]
- Sanchez, V.; Greis, K.D.; Sztul, E.; Britt, W.J. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: Characterization of a potential site of virus assembly. J. Virol. 2000, 74, 975–986. [Google Scholar] [CrossRef]
- Plachter, B.; Sinzger, C.; Jahn, G. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 1996, 46, 195–261. [Google Scholar]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [PubMed]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.; Lee, D.; Gelbmann, C.B.; Van Sciver, N.; Nawandar, D.M.; Kenney, S.C.; Kalejta, R.F. Human Cytomegalovirus Productively Replicates In Vitro in Undifferentiated Oral Epithelial Cells. J. Virol. 2018, 92, e00903-18. [Google Scholar] [CrossRef] [PubMed]
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 2011, 7, e1001256. [Google Scholar] [CrossRef]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 44. [Google Scholar] [CrossRef]
- Boeckh, M.; Nichols, W.G.; Papanicolaou, G.; Rubin, R.; Wingard, J.R.; Zaia, J. Cytomegalovirus in hematopoietic stem cell transplant recipients: Current status, known challenges, and future strategies. Biol. Blood Marrow Transplant. 2003, 9, 543–558. [Google Scholar] [CrossRef]
- Rafailidis, P.I.; Mourtzoukou, E.G.; Varbobitis, I.C.; Falagas, M.E. Severe cytomegalovirus infection in apparently immunocompetent patients: A systematic review. Virol. J. 2008, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Steininger, C. Clinical relevance of cytomegalovirus infection in patients with disorders of the immune system. Clin. Microbiol. Infect. 2007, 13, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.G.; Lurain, N.S.; Zuckerman, T.; Hoffman, R.; Satinger, J.; Honigman, A.; Saleh, N.; Robert, E.S.; Rowe, J.M.; Kra-Oz, Z. Emergence of late cytomegalovirus central nervous system disease in hematopoietic stem cell transplant recipients. Blood 2003, 101, 463–465. [Google Scholar] [CrossRef]
- Britt, W.J. Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. J. Virol. 2017, 91, e02392-16. [Google Scholar] [CrossRef]
- Buxmann, H.; Hamprecht, K.; Meyer-Wittkopf, M.; Friese, K. Primary Human Cytomegalovirus (HCMV) Infection in Pregnancy. Dtsch. Arztebl. Int. 2017, 114, 45–52. [Google Scholar] [CrossRef]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef]
- Hamilton, S.T.; van Zuylen, W.; Shand, A.; Scott, G.M.; Naing, Z.; Hall, B.; Craig, M.E.; Rawlinson, W.D. Prevention of congenital cytomegalovirus complications by maternal and neonatal treatments: A systematic review. Rev. Med. Virol. 2014, 24, 420–433. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Human cytomegalovirus tropism for endothelial/epithelial cells: Scientific background and clinical implications. Rev. Med. Virol. 2010, 20, 136–155. [Google Scholar] [CrossRef]
- Sia, I.G.; Patel, R. New strategies for prevention and therapy of cytomegalovirus infection and disease in solid-organ transplant recipients. Clin. Microbiol. Rev. 2000, 13, 83–121. [Google Scholar] [CrossRef]
- Tsutsui, Y. Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. 2009, 49, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J.; Prichard, M.N. New therapies for human cytomegalovirus infections. Antivir. Res. 2018, 159, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Craig, J.M.; Macauley, J.C.; Weller, T.H.; Wirth, P. Isolation of intranuclear inclusion producing agents from infants with illnesses resembling cytomegalic inclusion disease. Proc. Soc. Exp. Biol. Med. 1957, 94, 4–12. [Google Scholar] [PubMed]
- Riley, H.D., Jr. History of the cytomegalovirus. South. Med. J. 1997, 90, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.; Hartley, J.W.; Cramblett, H.G.; Mastrota, F.M. Detection of human salivary gland virus in the mouth and urine of children. Am. J. Hyg. 1958, 67, 57–65. [Google Scholar]
- Rowe, W.P.; Hartley, J.W.; Waterman, S.; Turner, H.C.; Huebner, R.J. Cytopathogenic agent resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc. Soc. Exp. Biol. Med. 1956, 92, 418–424. [Google Scholar]
- Smith, M.G. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc. Soc. Exp. Biol. Med. 1956, 92, 424–430. [Google Scholar] [CrossRef]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Goyette, N.; Gilbert, C.; Humar, A.; Covington, E. Clinical impact of ganciclovir-resistant cytomegalovirus infections in solid organ transplant patients. Transplant. Infect. Dis. 2005, 7, 166–170. [Google Scholar] [CrossRef]
- Danziger-Isakov, L.; Mark Baillie, G. Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clin. Transplant. 2009, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Härter, G.; Michel, D. Antiviral treatment of cytomegalovirus infection: An update. Expert Opin. Pharmacother. 2012, 13, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lischka, P.; Zimmermann, H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr. Opin. Pharmacol. 2008, 8, 541–548. [Google Scholar] [CrossRef]
- Shmueli, E.; Or, R.; Shapira, M.Y.; Resnick, I.B.; Caplan, O.; Bdolah-Abram, T.; Wolf, D.G. High rate of cytomegalovirus drug resistance among patients receiving preemptive antiviral treatment after haploidentical stem cell transplantation. J. Infect. Dis. 2014, 209, 557–561. [Google Scholar] [CrossRef]
- Chong, P.P.; Teiber, D.; Prokesch, B.C.; Arasaratnam, R.J.; Peltz, M.; Drazner, M.H.; Garg, S. Letermovir successfully used for secondary prophylaxis in a heart transplant recipient with ganciclovir-resistant cytomegalovirus syndrome (UL97 mutation). Transplant. Infect. Dis. 2018, 20, e12965. [Google Scholar] [CrossRef]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef]
- Lischka, P.; Hewlett, G.; Wunberg, T.; Baumeister, J.; Paulsen, D.; Goldner, T.; Ruebsamen-Schaeff, H.; Zimmermann, H. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 2010, 54, 1290–1297. [Google Scholar] [CrossRef]
- Wildum, S.; Zimmermann, H.; Lischka, P. In vitro drug combination studies of Letermovir (AIC246, MK-8228) with approved anti-human cytomegalovirus (HCMV) and anti-HIV compounds in inhibition of HCMV and HIV replication. Antimicrob. Agents Chemother. 2015, 59, 3140–3148. [Google Scholar] [CrossRef]
- Cherrier, L.; Nasar, A.; Goodlet, K.J.; Nailor, M.D.; Tokman, S.; Chou, S. Emergence of letermovir resistance in a lung transplant recipient with ganciclovir-resistant cytomegalovirus infection. Am. J. Transplant. 2018, 18, 3060–3064. [Google Scholar] [CrossRef]
- van Zeijl, M.; Fairhurst, J.; Baum, E.Z.; Sun, L.; Jones, T.R. The human cytomegalovirus UL97 protein is phosphorylated and a component of virions. Virology 1997, 231, 72–80. [Google Scholar] [CrossRef]
- Webel, R.; Hakki, M.; Prichard, M.N.; Rawlinson, W.D.; Marschall, M.; Chou, S. Differential properties of cytomegalovirus pUL97 kinase isoforms affect viral replication and maribavir susceptibility. J. Virol. 2014, 88, 4776–4785. [Google Scholar] [CrossRef] [PubMed]
- Webel, R.; Milbradt, J.; Auerochs, S.; Schregel, V.; Held, C.; Nobauer, K.; Razzazi-Fazeli, E.; Jardin, C.; Wittenberg, T.; Sticht, H.; et al. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 2011, 92, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Webel, R.; Solbak, S.M.; Held, C.; Milbradt, J.; Gross, A.; Eichler, J.; Wittenberg, T.; Jardin, C.; Sticht, H.; Fossen, T.; et al. Nuclear import of isoforms of the cytomegalovirus kinase pUL97 is mediated by differential activity of NLS1 and NLS2 both acting through classical importin-alpha binding. J. Gen. Virol. 2012, 93, 1756–1768. [Google Scholar] [CrossRef]
- Steingruber, M.; Socher, E.; Hutterer, C.; Webel, R.; Bergbrede, T.; Lenac, T.; Sticht, H.; Marschall, M. The Interaction between Cyclin B1 and Cytomegalovirus Protein Kinase pUL97 is Determined by an Active Kinase Domain. Viruses 2015, 7, 4582–4601. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Kalejta, R.F. Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 2009, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Gershburg, E.; Pagano, J.S. Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 2008, 1784, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Michel, D.; Mertens, T. The UL97 protein kinase of human cytomegalovirus and homologues in other herpesviruses: Impact on virus and host. Biochim. Biophys. Acta 2004, 1697, 169–180. [Google Scholar] [CrossRef]
- Marschall, M.; Stein-Gerlach, M.; Freitag, M.; Kupfer, R.; van Den Bogaard, M.; Stamminger, T. Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J. Gen. Virol. 2001, 82, 1439–1450. [Google Scholar] [CrossRef]
- Prichard, M.N. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 2009, 19, 215–229. [Google Scholar] [CrossRef]
- Marschall, M.; Feichtinger, S.; Milbradt, J. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 2011, 80, 69–101. [Google Scholar]
- Schregel, V.; Auerochs, S.; Jochmann, R.; Maurer, K.; Stamminger, T.; Marschall, M. Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Gen. Virol. 2007, 88, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Graf, L.; Webel, R.; Wagner, S.; Hamilton, S.T.; Rawlinson, W.D.; Sticht, H.; Marschall, M. The cyclin-dependent kinase ortholog pUL97 of human cytomegalovirus interacts with cyclins. Viruses 2013, 5, 3213–3230. [Google Scholar] [CrossRef] [PubMed]
- Steingruber, M.; Keller, L.; Socher, E.; Ferre, S.; Hesse, A.M.; Coute, Y.; Hahn, F.; Büscher, N.; Plachter, B.; Sticht, H.; et al. Cyclins B1, T1, and H differ in their molecular mode of interaction with cytomegalovirus protein kinase pUL97. J. Biol. Chem. 2019, 294, 6188–6203. [Google Scholar] [CrossRef] [PubMed]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed]
- Hertel, L.; Chou, S.; Mocarski, E.S. Viral and cell cycle-regulated kinases in cytomegalovirus-induced pseudomitosis and replication. PLoS Pathog. 2007, 3, e6. [Google Scholar] [CrossRef]
- Wolf, D.G.; Courcelle, C.T.; Prichard, M.N.; Mocarski, E.S. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. USA 2001, 98, 1895–1900. [Google Scholar] [CrossRef]
- Prichard, M.N.; Britt, W.J.; Daily, S.L.; Hartline, C.B.; Kern, E.R. Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J. Virol. 2005, 79, 15494–15502. [Google Scholar] [CrossRef]
- Prichard, M.N.; Gao, N.; Jairath, S.; Mulamba, G.; Krosky, P.; Coen, D.M.; Parker, B.O.; Pari, G.S. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 1999, 73, 5663–5670. [Google Scholar] [CrossRef]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27, e1934. [Google Scholar] [CrossRef]
- Romaker, D.; Schregel, V.; Maurer, K.; Auerochs, S.; Marzi, A.; Sticht, H.; Marschall, M. Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem. 2006, 49, 7044–7053. [Google Scholar] [CrossRef]
- Hutterer, C.; Hamilton, S.; Steingruber, M.; Zeittrager, I.; Bahsi, H.; Thuma, N.; Naing, Z.; Orfi, Z.; Orfi, L.; Socher, E.; et al. The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antivir. Res. 2016, 134, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.C.; Krosky, P.M.; Coen, D.M. Relationship between autophosphorylation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. J. Virol. 2002, 76, 11943–11952. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Perlman, D.H.; Shenk, T.; Terry, L.J. Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics 2015, 15, 2006–2022. [Google Scholar] [CrossRef]
- Michel, D.; Pavic, I.; Zimmermann, A.; Haupt, E.; Wunderlich, K.; Heuschmid, M.; Mertens, T. The UL97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. J. Virol. 1996, 70, 6340–6346. [Google Scholar] [CrossRef] [PubMed]
- Littler, E.; Stuart, A.D.; Chee, M.S. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 1992, 358, 160–162. [Google Scholar] [CrossRef]
- Sullivan, V.; Talarico, C.L.; Stanat, S.C.; Davis, M.; Coen, D.M.; Biron, K.K. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 359, 85. [Google Scholar] [CrossRef]
- Held, C.; Webel, R.; Palmisano, R.; Hutterer, C.; Marschall, M.; Wittenberg, T. Using multi-channel level sets to measure the cytoplasmic localization of HCMV pUL97 in GFP-B-gal fusion constructs. J. Virol. Methods 2014, 199, 61–67. [Google Scholar] [CrossRef]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. J. Virol. 2016, 90, 9483–9494. [Google Scholar] [CrossRef]
- Chou, S.; Ercolani, R.J.; Derakhchan, K. Antiviral activity of maribavir in combination with other drugs active against human cytomegalovirus. Antivir. Res. 2018, 157, 128–133. [Google Scholar] [CrossRef]
- Caviness, K.; Bughio, F.; Crawford, L.B.; Streblow, D.N.; Nelson, J.A.; Caposio, P.; Goodrum, F. Complex Interplay of the UL136 Isoforms Balances Cytomegalovirus Replication and Latency. mBio 2016, 7, e01986. [Google Scholar] [CrossRef]
- Sehl, J.; Portner, S.; Klupp, B.G.; Granzow, H.; Franzke, K.; Teifke, J.P.; Mettenleiter, T.C. Roles of the different isoforms of the pseudorabies virus protein kinase pUS3 in nuclear egress. J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Finkel, J.S.; Kamil, J.P.; Coen, D.M.; Culbertson, M.R.; Kalejta, R.F. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 2008, 320, 797–799. [Google Scholar] [CrossRef]
- Cunningham, C.; Gatherer, D.; Hilfrich, B.; Baluchova, K.; Dargan, D.J.; Thomson, M.; Griffiths, P.D.; Wilkinson, G.W.; Schulz, T.F.; Davison, A.J. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 2010, 91, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Weinberg, A.; Crumpacker, C.S.; Chou, S. Sequencing of cytomegalovirus UL97 gene for genotypic antiviral resistance testing. Antimicrob. Agents Chemother. 2001, 45, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Rechter, S.; Scott, G.M.; Eickhoff, J.; Zielke, K.; Auerochs, S.; Muller, R.; Stamminger, T.; Rawlinson, W.D.; Marschall, M. Cyclin-dependent Kinases Phosphorylate the Cytomegalovirus RNA Export Protein pUL69 and Modulate Its Nuclear Localization and Activity. J. Biol. Chem. 2009, 284, 8605–8613. [Google Scholar] [CrossRef]
- Thomas, M.; Rechter, S.; Milbradt, J.; Auerochs, S.; Muller, R.; Stamminger, T.; Marschall, M. Cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. J. Gen. Virol. 2009, 90, 567–578. [Google Scholar] [CrossRef]
- Feichtinger, S.; Stamminger, T.; Muller, R.; Graf, L.; Klebl, B.; Eickhoff, J.; Marschall, M. Recruitment of cyclin-dependent kinase 9 to nuclear compartments during cytomegalovirus late replication: Importance of an interaction between viral pUL69 and cyclin T1. J. Gen. Virol. 2011, 92, 1519–1531. [Google Scholar] [CrossRef]
- Graf, L.; Feichtinger, S.; Naing, Z.; Hutterer, C.; Milbradt, J.; Webel, R.; Wagner, S.; Scott, G.M.; Hamilton, S.T.; Rawlinson, W.D.; et al. New insight into the phosphorylation-regulated intranuclear localization of human cytomegalovirus pUL69 mediated by cyclin-dependent kinases (CDKs) and viral CDK orthologue pUL97. J. Gen. Virol. 2016, 97, 144–151. [Google Scholar] [CrossRef]
- Steingruber, M.; Kraut, A.; Socher, E.; Sticht, H.; Reichel, A.; Stamminger, T.; Amin, B.; Coute, Y.; Hutterer, C.; Marschall, M. Proteomic Interaction Patterns between Human Cyclins, the Cyclin-Dependent Kinase Ortholog pUL97 and Additional Cytomegalovirus Proteins. Viruses 2016, 8, 219. [Google Scholar] [CrossRef]
- König, P.; Buscher, N.; Steingruber, M.; Socher, E.; Sticht, H.; Tenzer, S.; Plachter, B.; Marschall, M. Dynamic regulatory interaction between cytomegalovirus major tegument protein pp65 and protein kinase pUL97 in intracellular compartments, dense bodies and virions. J. Gen. Virol. 2017, 98, 2850–2863. [Google Scholar] [CrossRef]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009, 5, e1000275. [Google Scholar] [CrossRef] [PubMed]
- Kamil, J.P.; Hume, A.J.; Jurak, I.; Munger, K.; Kalejta, R.F.; Coen, D.M. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 16823–16828. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Wandinger, S.K.; Wagner, S.; Muller, R.; Stamminger, T.; Zeittrager, I.; Godl, K.; Baumgartner, R.; Strobl, S.; Marschall, M. Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases Aurora A, ABL and AMPK. Antivir. Res. 2013, 99, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Sevvana, M.; Muller, Y.A.; Sticht, H.; Marschall, M. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J. Biol. Chem. 2012, 287, 24004–24016. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.B.; James, S.H.; Prichard, M.N. Human cytomegalovirus UL97 kinase alters the accumulation of CDK1. J. Gen. Virol. 2012, 93, 1743–1755. [Google Scholar] [CrossRef] [PubMed]
- Jault, F.M.; Jault, J.M.; Ruchti, F.; Fortunato, E.A.; Clark, C.; Corbeil, J.; Richman, D.D.; Spector, D.H. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 1995, 69, 6697–6704. [Google Scholar] [CrossRef]
- Kapasi, A.J.; Spector, D.H. Inhibition of the cyclin-dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl-terminal domain kinases cdk9 and cdk7 at the viral transcriptosome. J. Virol. 2008, 82, 394–407. [Google Scholar] [CrossRef]
- Sanchez, V.; McElroy, A.K.; Spector, D.H. Mechanisms governing maintenance of Cdk1/cyclin B1 kinase activity in cells infected with human cytomegalovirus. J. Virol. 2003, 77, 13214–13224. [Google Scholar] [CrossRef]
- Spector, D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015, 204, 409–419. [Google Scholar] [CrossRef]
- Tamrakar, S.; Kapasi, A.J.; Spector, D.H. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 2005, 79, 15477–15493. [Google Scholar] [CrossRef]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef]
- Becke, S.; Fabre-Mersseman, V.; Aue, S.; Auerochs, S.; Sedmak, T.; Wolfrum, U.; Strand, D.; Marschall, M.; Plachter, B.; Reyda, S. Modification of the major tegument protein pp65 of human cytomegalovirus inhibits virus growth and leads to the enhancement of a protein complex with pUL69 and pUL97 in infected cells. J. Gen. Virol. 2010, 91, 2531–2541. [Google Scholar] [CrossRef]
- Dell’Oste, V.; Gatti, D.; Gugliesi, F.; De Andrea, M.; Bawadekar, M.; Lo Cigno, I.; Biolatti, M.; Vallino, M.; Marschall, M.; Gariglio, M.; et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 2014, 88, 6970–6982. [Google Scholar] [CrossRef]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell Proteom. 2014, 13, 2132–2146. [Google Scholar] [CrossRef]
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 2015, 89, 523–534. [Google Scholar] [CrossRef]
- Baek, M.C.; Krosky, P.M.; Pearson, A.; Coen, D.M. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 2004, 324, 184–193. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Matsumura, T.; Roizman, B.; Hirai, K. Cellular elongation factor 1delta is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 1999, 73, 4456–4460. [Google Scholar] [CrossRef]
- Bigley, T.M.; Reitsma, J.M.; Mirza, S.P.; Terhune, S.S. Human cytomegalovirus pUL97 regulates the viral major immediate early promoter by phosphorylation-mediated disruption of histone deacetylase 1 binding. J. Virol. 2013, 87, 7393–7408. [Google Scholar] [CrossRef]
- Iwahori, S.; Umana, A.C.; VanDeusen, H.R.; Kalejta, R.F. Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J. Biol. Chem. 2017, 292, 6583–6599. [Google Scholar] [CrossRef]
- Krosky, P.M.; Baek, M.C.; Jahng, W.J.; Barrera, I.; Harvey, R.J.; Biron, K.K.; Coen, D.M.; Sethna, P.B. The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J. Virol. 2003, 77, 7720–7727. [Google Scholar] [CrossRef] [PubMed]
- Businger, R.; Deutschmann, J.; Gruska, I.; Milbradt, J.; Wiebusch, L.; Gramberg, T.; Schindler, M. Human cytomegalovirus overcomes SAMHD1 restriction in macrophages via pUL97. Nat. Microbiol. 2019, 4, 2260–2272. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Kalejta, R.F. Phosphorylation of transcriptional regulators in the retinoblastoma protein pathway by UL97, the viral cyclin-dependent kinase encoded by human cytomegalovirus. Virology 2017, 512, 95–103. [Google Scholar] [CrossRef]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; von Einem, J.; Marschall, M.; Plachter, B.; Gariglio, M.; De Andrea, M.; Landolfo, S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J. Virol. 2016, 90, 8238–8250. [Google Scholar] [CrossRef]
- Landolfo, S.; De Andrea, M.; Dell’Oste, V.; Gugliesi, F. Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World J. Virol. 2016, 5, 87–96. [Google Scholar] [CrossRef]
- Marschall, M.; Stein-Gerlach, M.; Freitag, M.; Kupfer, R.; van den Bogaard, M.; Stamminger, T. Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. J. Gen. Virol. 2002, 83, 1013–1023. [Google Scholar] [CrossRef]
- He, Z.; He, Y.S.; Kim, Y.; Chu, L.; Ohmstede, C.; Biron, K.K.; Coen, D.M. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 1997, 71, 405–411. [Google Scholar] [CrossRef]
- Mett, H.; Holscher, K.; Degen, H.; Esdar, C.; De Neumann, B.F.; Flicke, B.; Freudenreich, T.; Holzer, G.; Schinzel, S.; Stamminger, T.; et al. Identification of inhibitors for a virally encoded protein kinase by 2 different screening systems: In vitro kinase assay and in-cell activity assay. J. Biomol. Screen. 2005, 10, 36–45. [Google Scholar] [CrossRef]
- Zimmermann, A.; Michel, D.; Pavic, I.; Hampl, W.; Luske, A.; Neyts, J.; De Clercq, E.; Mertens, T. Phosphorylation of aciclovir, ganciclovir, penciclovir and S2242 by the cytomegalovirus UL97 protein: A quantitative analysis using recombinant vaccinia viruses. Antivir. Res. 1997, 36, 35–42. [Google Scholar] [CrossRef]
- Talarico, C.L.; Burnette, T.C.; Miller, W.H.; Smith, S.L.; Davis, M.G.; Stanat, S.C.; Ng, T.I.; He, Z.; Coen, D.M.; Roizman, B.; et al. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein. Antimicrob. Agents Chemother. 1999, 43, 1941–1946. [Google Scholar] [CrossRef]
- Chou, S.; Wechel, L.C.; Marousek, G.I. Cytomegalovirus UL97 kinase mutations that confer maribavir resistance. J. Infect. Dis. 2007, 196, 91–94. [Google Scholar] [CrossRef]
- Scott, G.M.; Isaacs, M.A.; Zeng, F.; Kesson, A.M.; Rawlinson, W.D. Cytomegalovirus antiviral resistance associated with treatment induced UL97 (protein kinase) and UL54 (DNA polymerase) mutations. J. Med. Virol. 2004, 74, 85–93. [Google Scholar] [CrossRef]
- Wolf, D.G.; Honigman, A.; Lazarovits, J.; Tavor, E.; Panet, A. Characterization of the human cytomegalovirus UL97 gene product as a virion-associated protein kinase. Arch. Virol. 1998, 143, 1223–1232. [Google Scholar] [CrossRef]
- Chevillotte, M.; Landwehr, S.; Linta, L.; Frascaroli, G.; Luske, A.; Buser, C.; Mertens, T.; von Einem, J. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J. Virol. 2009, 83, 2480–2490. [Google Scholar] [CrossRef]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human Cytomegalovirus Nuclear Capsids Associate with the Core Nuclear Egress Complex and the Viral Protein Kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef]
- Herget, T.; Marschall, M. Recent developments in anti-herpesviral combination therapy based on protein kinase inhibitors. In New Concepts of Antiviral Therapy; Springer: Boston, MA, USA, 2006; pp. 351–371. [Google Scholar]
- Zimmermann, A.; Wilts, H.; Lenhardt, M.; Hahn, M.; Mertens, T. Indolocarbazoles exhibit strong antiviral activity against human cytomegalovirus and are potent inhibitors of the pUL97 protein kinase. Antivir. Res. 2000, 48, 49–60. [Google Scholar] [CrossRef]
- Slater, M.J.; Baxter, R.; Bonser, R.W.; Cockerill, S.; Gohil, K.; Parry, N.; Robinson, E.; Randall, R.; Yeates, C.; Snowden, W.; et al. Synthesis of N-alkyl substituted indolocarbazoles as potent inhibitors of human cytomegalovirus replication. Bioorg. Med. Chem. Lett. 2001, 11, 1993–1995. [Google Scholar] [CrossRef]
- Marschall, M.; Freitag, M.; Suchy, P.; Romaker, D.; Kupfer, R.; Hanke, M.; Stamminger, T. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 2003, 311, 60–71. [Google Scholar] [CrossRef]
- Schleiss, M.; Eickhoff, J.; Auerochs, S.; Leis, M.; Abele, S.; Rechter, S.; Choi, Y.; Anderson, J.; Scott, G.; Rawlinson, W.; et al. Protein kinase inhibitors of the quinazoline class exert anti-cytomegaloviral activity in vitro and in vivo. Antivir. Res. 2008, 79, 49–61. [Google Scholar] [CrossRef]
- Chou, S.; Marousek, G.I. Maribavir antagonizes the antiviral action of ganciclovir on human cytomegalovirus. Antimicrob. Agents Chemother. 2006, 50, 3470–3472. [Google Scholar] [CrossRef]
- Cazorla-Vazquez, S.; Steingruber, M.; Marschall, M.; Engel, F.B. Human cytomegaloviral multifunctional protein kinase pUL97 impairs zebrafish embryonic development and increases mortality. Sci. Rep. 2019, 9, 7219. [Google Scholar] [CrossRef] [PubMed]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Hage, S.; Kraut, A.; Hesse, A.M.; Amin, B.; Sonnewald, U.; Coute, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef]
- Sonntag, E.; Hamilton, S.T.; Bahsi, H.; Wagner, S.; Jonjic, S.; Rawlinson, W.D.; Marschall, M.; Milbradt, J. Erratum to Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 2016, 97, 2461. [Google Scholar] [CrossRef]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kato, K.; Tanaka, M.; Kanamori, M.; Nishiyama, Y.; Yamanashi, Y. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 2003, 77, 2359–2368. [Google Scholar] [CrossRef]
- Ruiz-Carrascoso, G.; Romero-Gomez, M.P.; Plaza, D.; Mingorance, J. Rapid detection and quantitation of ganciclovir resistance in cytomegalovirus quasispecies. J. Med. Virol. 2013, 85, 1250–1257. [Google Scholar] [CrossRef]
- Chou, S. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 2008, 18, 233–246. [Google Scholar] [CrossRef]
- Chou, S.; Van Wechel, L.C.; Marousek, G.I. Effect of cell culture conditions on the anticytomegalovirus activity of maribavir. Antimicrob. Agents Chemother. 2006, 50, 2557–2559. [Google Scholar] [CrossRef]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef]
- Muller, Y.A.; Häge, S.; Alkhashrom, S.; Höllriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical β- and γ-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef]
- Dal Monte, P.; Pignatelli, S.; Zini, N.; Maraldi, N.M.; Perret, E.; Prevost, M.C.; Landini, M.P. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J. Gen. Virol. 2002, 83, 1005–1012. [Google Scholar] [CrossRef]
- Lischka, P.; Rosorius, O.; Trommer, E.; Stamminger, T. A novel transferable nuclear export signal mediates CRM1-independent nucleocytoplasmic shuttling of the human cytomegalovirus transactivator protein pUL69. EMBO J. 2001, 20, 7271–7283. [Google Scholar] [CrossRef]
- Thomas, M.; Müller, R.; Horn, G.; Bogdanow, B.; Imami, K.; Milbradt, J.; Steingruber, M.; Marschall, M.; Schilling, E.M.; Fossen, T.; et al. Phosphosite analysis of the cytomegaloviral mRNA export factor pUL69 reveals serines with critical importance for recruitment of cellular proteins Pin1 and UAP56/URH49. J. Virol. 2020, 94, e02151-19. [Google Scholar] [CrossRef]
- Sharma, M.; Kamil, J.P.; Coen, D.M. Preparation of the Human Cytomegalovirus Nuclear Egress Complex and Associated Proteins. Methods Enzymol. 2016, 569, 517–526. [Google Scholar]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef]
- Deutschmann, J.; Schneider, A.; Gruska, I.; Vetter, B.; Thomas, D.; Kießling, M.; Wittmann, S.; Herrmann, A.; Schindler, M.; Milbradt, J.; et al. A viral kinase counteracts in vivo restriction of murine cytomegalovirus by SAMHD1. Nat. Microbiol. 2019, 4, 2273–2284. [Google Scholar] [CrossRef]
- Sanchez, V.; Spector, D.H. Cyclin-dependent kinase activity is required for efficient expression and posttranslational modification of human cytomegalovirus proteins and for production of extracellular particles. J. Virol. 2006, 80, 5886–5896. [Google Scholar] [CrossRef]
- Adams, P.D. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 2001, 1471, M123–M133. [Google Scholar] [CrossRef]
- Prichard, M.N.; Sztul, E.; Daily, S.L.; Perry, A.L.; Frederick, S.L.; Gill, R.B.; Hartline, C.B.; Streblow, D.N.; Varnum, S.M.; Smith, R.D.; et al. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 2008, 82, 5054–5067. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Chiu, H.C.; Huang, W.R.; Liao, T.L.; Chi, P.I.; Nielsen, B.L.; Liu, J.H.; Liu, H.J. Mechanistic insights into avian reovirus p17-modulated suppression of cell cycle CDK-cyclin complexes and enhancement of p53 and cyclin H interaction. J. Biol. Chem. 2018, 293, 12542–12562. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Gautier, J.; Minshull, J.; Lohka, M.; Glotzer, M.; Hunt, T.; Maller, J.L. Cyclin is a component of maturation-promoting factor from Xenopus. Cell 1990, 60, 487–494. [Google Scholar] [CrossRef]
- Lolli, G.; Lowe, E.D.; Brown, N.R.; Johnson, L.N. The crystal structure of human CDK7 and its protein recognition properties. Structure 2004, 12, 2067–2079. [Google Scholar] [CrossRef]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Shinya, N.; Iwamatsu, A.; Nishida, E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001, 410, 215–220. [Google Scholar] [CrossRef]
- Yuan, J.; Eckerdt, F.; Bereiter-Hahn, J.; Kurunci-Csacsko, E.; Kaufmann, M.; Strebhardt, K. Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene 2002, 21, 8282–8292. [Google Scholar] [CrossRef]
- Bigley, T.M.; Reitsma, J.M.; Terhune, S.S. Antagonistic Relationship between Human Cytomegalovirus pUL27 and pUL97 Activities during Infection. J. Virol. 2015, 89, 10230–10246. [Google Scholar] [CrossRef]
- Garrett, S.; Barton, W.A.; Knights, R.; Jin, P.; Morgan, D.O.; Fisher, R.P. Reciprocal activation by cyclin-dependent kinases 2 and 7 is directed by substrate specificity determinants outside the T loop. Mol. Cell. Biol. 2001, 21, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sharp, P.A. Positive transcription elongation factor B phosphorylates hSPT5 and RNA polymerase II carboxyl-terminal domain independently of cyclin-dependent kinase-activating kinase. J. Biol. Chem. 2001, 276, 12317–12323. [Google Scholar] [CrossRef]
- Martinez, A.M.; Afshar, M.; Martin, F.; Cavadore, J.C.; Labbe, J.C.; Doree, M. Dual phosphorylation of the T-loop in cdk7: Its role in controlling cyclin H binding and CAK activity. EMBO J. 1997, 16, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Rice, A.P. Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. J. Cell. Physiol. 2012, 227, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef]
- Timofeev, O.; Cizmecioglu, O.; Settele, F.; Kempf, T.; Hoffmann, I. Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J. Biol. Chem. 2010, 285, 16978–16990. [Google Scholar] [CrossRef]
- Lees, J.A.; Buchkovich, K.J.; Marshak, D.R.; Anderson, C.W.; Harlow, E. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J. 1991, 10, 4279–4290. [Google Scholar] [CrossRef]
- Simone, C.; Bagella, L.; Bellan, C.; Giordano, A. Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene 2002, 21, 4158–4165. [Google Scholar] [CrossRef]
- Ko, L.J.; Shieh, S.Y.; Chen, X.; Jayaraman, L.; Tamai, K.; Taya, Y.; Prives, C.; Pan, Z.Q. p53 is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner. Mol. Cell. Biol. 1997, 17, 7220–7229. [Google Scholar] [CrossRef]
- Nantajit, D.; Fan, M.; Duru, N.; Wen, Y.; Reed, J.C.; Li, J.J. Cyclin B1/Cdk1 phosphorylation of mitochondrial p53 induces anti-apoptotic response. PLoS ONE 2010, 5, e12341. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Gartel, A.L. CDK9 phosphorylates p53 on serine residues 33, 315 and 392. Cell Cycle 2006, 5, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.; McKeon, F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579–589. [Google Scholar] [CrossRef]
- Ward, G.E.; Kirschner, M.W. Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 1990, 61, 561–577. [Google Scholar] [CrossRef]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef]
- Lolli, G. Binding to DNA of the RNA-polymerase II C-terminal domain allows discrimination between Cdk7 and Cdk9 phosphorylation. Nucleic Acids Res. 2009, 37, 1260–1268. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadao, A.L.; Laguette, N.; Benkirane, M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef]
- White, T.E.; Brandariz-Nunez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef]
- Zhang, K.; Lv, D.-W.; Li, R. Conserved Herpesvirus Protein Kinases Target SAMHD1 to Facilitate Virus Replication. Cell Rep. 2019, 28, 449–459. [Google Scholar] [CrossRef]
- Gentry, B.G.; Gentry, S.N.; Jackson, T.L.; Zemlicka, J.; Drach, J.C. Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochem. Pharmacol. 2011, 81, 43–49. [Google Scholar] [CrossRef]
- Biron, K.K.; Harvey, R.J.; Chamberlain, S.C.; Good, S.S.; Smith, A.A., 3rd; Davis, M.G.; Talarico, C.L.; Miller, W.H.; Ferris, R.; Dornsife, R.E.; et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action. Antimicrob. Agents Chemother. 2002, 46, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Strojan, H.; Kiener, R.; Wangen, C.; Sonntag, E.; Muller, R.; Zeittrager, I.; Wagner, S.; Stamminger, T.; Milbradt, J.; et al. Differential upregulation of host cell protein kinases by the replication of alpha-, beta- and gamma-herpesviruses provides a signature of virus-specific signalling. J. Gen. Virol. 2020, 101, 284–289. [Google Scholar] [CrossRef]
- Mercorelli, B.; Sinigalia, E.; Loregian, A.; Palu, G. Human cytomegalovirus DNA replication: Antiviral targets and drugs. Rev. Med. Virol. 2008, 18, 177–210. [Google Scholar] [CrossRef] [PubMed]
- Herget, T.; Freitag, M.; Morbitzer, M.; Kupfer, R.; Stamminger, T.; Marschall, M. Novel chemical class of pUL97 protein kinase-specific inhibitors with strong anticytomegaloviral activity. Antimicrob. Agents Chemother. 2004, 48, 4154–4162. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Ercolani, R.J.; Marousek, G.; Bowlin, T.L. Cytomegalovirus UL97 kinase catalytic domain mutations that confer multidrug resistance. Antimicrob. Agents Chemother. 2013, 57, 3375–3379. [Google Scholar] [CrossRef] [PubMed]
- Biron, K.K. Maribavir: A Promising New Antiherpes Therapeutic Agent. In New Concepts of Antiviral Therapy; Springer: Boston, MA, USA, 2006. [Google Scholar]
- Koszalka, G.W.; Johnson, N.W.; Good, S.S.; Boyd, L.; Chamberlain, S.C.; Townsend, L.B.; Drach, J.C.; Biron, K.K. Preclinical and toxicology studies of 1263W94, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 2002, 46, 2373–2380. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Aberg, J.A.; Wang, L.H.; Wire, M.B.; Miner, R.; Snowden, W.; Talarico, C.L.; Shaw, S.; Jacobson, M.A.; Drew, W.L. Phase I dose escalation trial evaluating the pharmacokinetics, anti-human cytomegalovirus (HCMV) activity, and safety of 1263W94 in human immunodeficiency virus-infected men with asymptomatic HCMV shedding. Antimicrob. Agents Chemother. 2002, 46, 2969–2976. [Google Scholar] [CrossRef]
- Ma, J.D.; Nafziger, A.N.; Villano, S.A.; Gaedigk, A.; Bertino, J.S., Jr. Maribavir pharmacokinetics and the effects of multiple-dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N-acetyltransferase-2, and xanthine oxidase activities in healthy adults. Antimicrob. Agents Chemother. 2006, 50, 1130–1135. [Google Scholar] [CrossRef]
- Marty, F.M.; Boeckh, M. Maribavir and human cytomegalovirus-what happened in the clinical trials and why might the drug have failed? Curr. Opin. Virol. 2011, 1, 555–562. [Google Scholar] [CrossRef]
- Evers, D.L.; Komazin, G.; Shin, D.; Hwang, D.D.; Townsend, L.B.; Drach, J.C. Interactions among antiviral drugs acting late in the replication cycle of human cytomegalovirus. Antivir. Res. 2002, 56, 61–72. [Google Scholar] [CrossRef]
- James, S.H.; Hartline, C.B.; Harden, E.A.; Driebe, E.M.; Schupp, J.M.; Engelthaler, D.M.; Keim, P.S.; Bowlin, T.L.; Kern, E.R.; Prichard, M.N. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 2011, 55, 4682–4691. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Waldemer, R.H.; Senters, A.E.; Michels, K.S.; Kemble, G.W.; Miner, R.C.; Drew, W.L. Cytomegalovirus UL97 phosphotransferase mutations that affect susceptibility to ganciclovir. J. Infect. Dis. 2002, 185, 162–169. [Google Scholar] [CrossRef] [PubMed]
- McSharry, J.J.; McDonough, A.; Olson, B.; Talarico, C.; Davis, M.; Biron, K.K. Inhibition of ganciclovir-susceptible and -resistant human cytomegalovirus clinical isolates by the benzimidazole L-riboside 1263W94. Clin. Diagn. Lab. Immunol. 2001, 8, 1279–1281. [Google Scholar] [CrossRef]
- Williams, S.L.; Hartline, C.B.; Kushner, N.L.; Harden, E.A.; Bidanset, D.J.; Drach, J.C.; Townsend, L.B.; Underwood, M.R.; Biron, K.K.; Kern, E.R. In vitro activities of benzimidazole D- and L-ribonucleosides against herpesviruses. Antimicrob. Agents Chemother. 2003, 47, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Marousek, G.I.; Senters, A.E.; Davis, M.G.; Biron, K.K. Mutations in the human cytomegalovirus UL27 gene that confer resistance to maribavir. J. Virol. 2004, 78, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Komazin, G.; Townsend, L.B.; Drach, J.C. Role of a mutation in human cytomegalovirus gene UL104 in resistance to benzimidazole ribonucleosides. J. Virol. 2004, 78, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Quenelle, D.C.; Bidanset, D.J.; Komazin, G.; Chou, S.; Drach, J.C.; Kern, E.R. Human cytomegalovirus UL27 is not required for viral replication in human tissue implanted in SCID mice. Virol. J. 2006, 3, 18. [Google Scholar] [CrossRef]
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms8040515