On various La
XSr
1−xCoO
3−δ as OER catalysts, a general strategy was demonstrated for steering the two mechanisms, AEM and LOM. By delicately controlling the oxygen defect contents, the dominant OER mechanism can be arbitrarily transformed between AEM-LOM-AEM accompanied by a volcano-type activity variation trend. Experimental and computational evidence explicitly revealed that the phenomenon is due to the fact that the increased oxygen defects alter the lattice oxygen activity with a volcano-type trend and preserve the Co
0 state for preferably the OER [
173].
For NdNiO
3, the link between structural anisotropy and the OER catalytic activity was established by DFT calculation. The NdNiO
3 with (100), (110), and (111) orientations display similar oxidative states and metal–oxygen covalence characteristics, but distinct OER activities in experimental results were in the order of (100) > (110) > (111). DFT results confirm that film orientation is a critical determinant of the reaction mechanism. The OER on the (100)-surface favors proceeding via a LOM. In contrast, the reaction on (110)- and (111)-surfaces followed the AEM. The anisotropic oxygen vacancy formation energy and stability are strongly correlated to the reaction mechanism and performance [
174]. On LaNiO
3 epitaxial thin films, electrochemical scanning tunneling microscopy (EC-STM) was used to directly observe structural dynamics during the OER. Based on the comparison of dynamic topographical changes in different compositions, reconstruction of surface morphology originating from the transition of the Ni species on the surface termination during the OER was proposed [
175]. The change in surface topography was induced by Ni(OH)
2/NiOOH redox transformation by quantifying STM images [
175].
On La
1−xNiO
3 perovskite electrocatalysts, direct O–O coupling promoted the OER activity at the interfacial active sites for decorated Ag nanoparticles. The theoretical calculation revealed that oxygen evolution via the dual-site mechanism with direct O–O coupling becomes more favorable than that via the conventional AEM. At x = 0.05, the electrocatalyst showed 20 times higher mass activity than that of the IrO
2 electrocatalyst, and the activity increased to 74 times after an accelerated durability test [
176].
Ca
2−xIrO
4 nanocrystals exhibited a very high stability of about 62 times that of benchmark IrO
2. Lattice resolution images and surface-sensitive spectroscopies demonstrated the Ir-rich surface layer with high relative content of Ir
5+ sites, which is responsible for the high activity and long-term stability. Combining operando IR spectroscopy with the XAS method, key intermediates of Ir
6+ = O and Ir
6+OO
− on Ir-based oxides electrocatalysts were observed, and they were stable even just from 1.3 V vs. RHE. DFT calculations indicated that the catalytic activity of Ca
2IrO
4 is enhanced remarkably after leaching the surface Ca ions because Ir = O and IrOO
− intermediates can be stabilized on positively charged active sites of the Ir-rich surface layer [
63]. Layered perovskite Sr
2IrO
4 was chemically exfoliated into protonated colloidal nanosheets with an undamaged perovskite framework. This OER catalyst exhibited about 10 times higher activity than the IrO
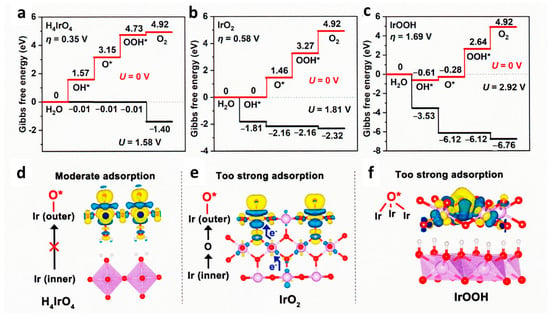
2 catalyst film. As shown in
Figure 11, DFT calculation indicated that electrons from the inner Ir atom to the surface were observed in IrO
2 (e) but not in the case of perovskite nanosheet (d), then the free energy of O* is moderate as shown in (a) compared with the case of IrO
2 (b). Thus, the structural hydroxyl groups on the surface of protonated nanosheets participate in the catalytic cycle [
177].
Figure 11. DFT calculation of HION, IrO
2, and IrOOH. (
a–
c) Gibbs free energy diagrams of H
4IrO
4, IrO
2, and IrOOH for OER at Ir sites. (
d–
f) Charge density difference induced by oxygen adsorption of H
4IrO
4, IrO
2, and IrOOH at Ir sites. Reproduced from
ACS Catal.
2022 [
177] with permission from the American Chemical Society.
7. Transition Metal (TM) Compounds
Multicomponent transition metal oxides and (oxy)hydroxides are the most promising OER catalysts due to their low cost, adjustable structure, high electrocatalytic activity, and outstanding durability. Co-, Ni-, and Fe-based OER catalysts have been considered to be potential candidates to replace noble metals, especially for electrocatalysts, due to their tunable 3d electron configuration and spin state, versatility in terms of crystal and electronic structures, as well as abundance in nature [
23]. The latest advances in the rational design of the related OER electrocatalysts and the modulation of the electronic structure of active sites were comprehensively summarized, besides a brief overview of the mechanisms of OER and the theory and calculation criteria [
178].
Wang et al. reviewed the fundamental understanding of the electronic structure of low-cost TM oxide-based catalysts for electrochemical OER, and its relationship with the catalytic activity and the reaction mechanism was discussed [
179]. Feng et al. reviewed the relationship between TMs and OER catalyst activity, and then the mechanism of synthesis strategy in different types of TMs-based catalysts was summarized [
180]. Guo et al. reviewed the state-of-the-art amorphous transition metal-based OER electrocatalysts, involving oxides, hydroxides, sulfides, phosphides, borides, and their composites, and then the practical application and theoretical modeling of the OER mechanisms in the OER were presented [
181]. Though transition metal phosphides often exhibit excellent HER activity, the OER catalytic performance is not outstanding. Huang et al. reviewed the strategies for preparing highly active OER catalysts of transition metal phosphides [
182].
The early transition metals (Ti, V, and Cr) can form very stable M=O units, while the late transition metals (Ni and Cu) can only theoretically form unstable M=O structures. On the other hand, for Mn, Fe, and Co, the metal-oxo motif switches between two valence tautomers in the form of Mn
+1 = O
2− and Mn–O•
−. The former with an electrophilic oxygen atom can proceed via the acid–base WNA pathway to form the O–O bond, whereas the latter favors the oxygen radical coupling pathway for O–O bond formation [
183].
7.1. CoOx
CoOOH was selected as the OER co-catalyst of aluminum-doped strontium titanate (SrTiO:Al) photocatalyst to attain almost unity in the internal quantum efficiency of UV-induced water splitting with Rh/Cr HER co-catalyst [
184]. The recent progress of Co
3O
4-based electrocatalytic materials for the acidic OER was presented with particular reference to the catalytic mechanism and guidelines for the design principles from both experimental and theoretical perspectives [
185]. Afterward, emerging strategies were outlined to improve the catalytic performance of Co
3O
4-based acidic OER catalysts, including phase engineering, component regulation with doping, composite with carbon-based materials, and multi-phase hybridization [
185].
For the application of Co oxides to photocatalysts, operando XPS measurements were performed. The catalyst undergoes chemical–structural transformations as a function of the applied anodic potential, with complete conversion of the Co(OH)
2 and partial conversion of the spinel Co
3O
4 phases to cobalt oxyhydroxide, CoO(OH), under precatalytic electrochemical conditions. This interpretation revealed that the presence of Co(OH)
2 enhances catalytic activity by promoting transformations to CoO(OH) [
186]. To study the mechanism of OER on CoO(OH), operando X-ray absorption and Raman spectroscopy revealed that a Co(IV) species, CoO
2, is the dominating resting state of the catalyst. Oxygen isotope exchange experiments showed that a cobalt superoxide species is an active intermediate in the OER. This intermediate is formed concurrently with the oxidation of CoO(OH) to CoO
2. Combining spectroscopic and electrokinetic data, the rate-determining step of the OER was identified as the release of dioxygen from the superoxide intermediate [
92].
By using a water-in-salt electrolyte, the water activity was systematically tuned and the mechanism as a function of applied potentials in water electrolysis was probed. The mechanism is sensitive to the applied potential. The Co-OO-Co bond forms via an intramolecular oxygen coupling mechanism at low potentials, whereas it proceeds through a WNA mechanism by forming Co-OOH at high potentials [
187].
The morphology-dependent analysis for well-defined crystalline CoO(OH) revealed that the active sites are exclusively located at lateral facets rather than basal facets. Theoretical calculations show that the coordinately unsaturated Co sites of lateral facets upshift the O 2p-band center closer to the Fermi level, thereby enhancing the covalency of Co-O bonds to yield the reactivity [
188]. The sequential oxidation kinetics with Co
3O
4 nanoparticles involving multi-active sites for water oxidation in the OER catalytic cycle were resolved by applying quasi-operando transient absorption spectroscopy to a typical photosensitization with Ru-dye and sacrificial electron donor. The Co
IV intermediate distribution plays a determining role in OER activity and results in the slow overall OER kinetics [
189]. The redox process between Co
III and Co
IV species does not follow a proton-coupled electron transfer mechanism that is thought to be common prior to the OER, but it involves a proton-decoupled electron transfer, clarified by isotope labeling experiments and in situ electrostatic modulation [
190].
An oxygen vacancy (Vo)-rich environment facilitates the reconstruction of Co
3O
4 to the Co(OH)
2 intermediate with proton vacancies (Co
IIO
x(OH)
y), which is favorable for the formation of the active species of CoO(OH). Correlative operando Raman spectra characterizations and electrokinetic analyses indicated that a moderate Vo density can switch the O–O bond formation pathway, from a WNA to an INA pathway, which is more kinetically favorable for water oxidation [
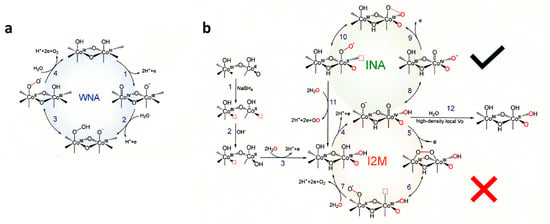
191]. As shown in
Figure 12b with O vacancy, at step 3, three protons and one electron are removed to form Co-O(OH). At step 4, Co
III sites of Co-O(OH) are oxidized to Co
IV which can be deprotonated (step 8) by hole attack oxo ligand Co
IV = O forms a Co-O-O triangle (step 9), and then becomes Co
II-OO• (step 10). At the next oxidation (step 10), O
2 is released and Co
II back to Co
III with the coordination of water. The I2M process was excluded based on the experimental results of using the H
218O isotope [
191].
Figure 12. Proposed reaction mechanisms of the OER for (
a) Co
3O
4 and (
b) Vo-Co
3O
4. Oxygen vacancy (Vo) prepared by NaBH
4 reduction was represented by the red dotted boxes. WNA = water nucleophilic attack, INA = intramolecular nucleophilic attack, and I2M = interaction between the two metal-O units. Reproduced from
ACS Catal. 2023 [
191] with permission from the American Chemical Society.
Amorphous CoO(OH) layer architecture was loaded onto the surface of TiO
2. Tafel analysis, EIS, and CV methods showed that the carrier transfer barrier within the electrode and the transition of Co
IIIO(OH) to Co
IVO(OH) have the dominating effects on the photoelectrochemical performance. Theoretical calculation revealed that the interface between the CoO(OH) and TiO
2 improves the electronic transfer ability among Co sites [
192]. Amorphous CoO(OH) layers are electrochemically synthesized on the surface of various cobalt sulfides CoS
α and found to decrease the intermolecular energy gap. The decrease in the energy gap accelerates the formation of OER-active high-valent Co
IV species [
193].
7.2. NiOx
For the nascent ultra-small nickel oxyhydroxide (NiOOH) particles (<3 nm), the thermodynamics of Ni dissolution was calculated by using first-principles theory at a near-neutral pH range, and the mechanism of OER on the γ-NiOOH surface was clarified. It was concluded that (i) ∼4% Ni cations on the surface of γ-NiOOH dissolve at pH = 7 and 1.73 V vs. RHE; (ii) on the pristine γ-NiOOH surface, OER proceeds via the “lattice peroxide” mechanism (*H
2O → *OH → *O–O
lattH* → O–O
latt → O
2) with an overpotential of 0.70 V; (iii) in the presence of Ni cationic vacancies, OER proceeds via the “hydroperoxide” mechanism (*OH + *H
2O → *2OH → *OOH → O
2) with an overpotential of 0.40 V [
196].
For NiOOH-based materials, light-triggered reversible geometric conversion between octahedron (NiO
6) and square planar (NiO
4) was proposed. The unit cell was undergone to achieve electronic states with alternative metal and oxygen characters throughout the oxygen evolution process. Utilizing this electron transfer pathway can bypass the potential limiting steps, that is, O–O bonding in the AEM and deprotonation in the LOM. As a result, the electrocatalysts that operate through this route showed superior activity compared with previously reported electrocatalysts [
197,
198].
By incorporating Fe and V into Ni(OH)
2 lattices, OER activity was improved. X-ray photoelectron/absorption spectroscopies revealed the synergistic interaction between Fe/V dopants and Ni in the host matrix, which subtly modulates local coordination environments and electronic structures of the Fe/V/Ni cations. Further, in situ XAS analyses manifested contraction of metal–oxygen bond lengths in the activated catalyst, with a short V–O bond distance. DFT calculations indicated that the V site of the Fe/V co-doped NiOOH gave near-optimal binding energies of OER intermediates and had lower overpotential compared with Ni and Fe sites [
199]. A series of Mn-, Co-, Fe-, and Zn-doped nickel oxides were investigated by using operando UV−vis spectroscopy coupled with time-resolved stepped potential spectroelectrochemistry. The Ni
2+/Ni
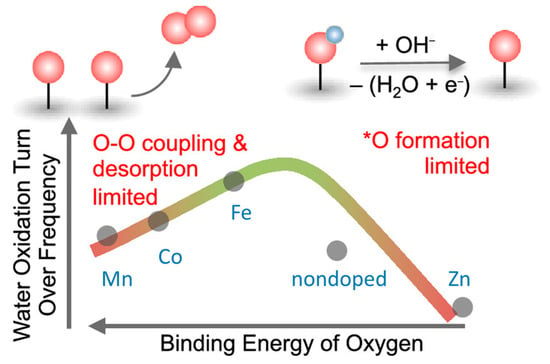
3+ redox peak potential was found to shift anodically from Mn- < Co- < Fe- < Zn-doped samples, suggesting a decrease in oxygen binding energetics from Mn- to Zn-doped samples. The OER kinetics had a second-order dependence on the density of these oxidized species, suggesting a chemical rate-determining step involving the coupling of two oxo species. The intrinsic turnover frequency per oxidized species exhibits a volcano trend with the binding energy of oxygen on the Ni site, having a maximum activity for the Fe-doped sample as shown in
Figure 13. For Ni centers that bind oxygen too strongly (Mn- and Co-doped oxides), OER kinetics is limited by O–O coupling and oxygen desorption, while for Ni centers that bind oxygen too weakly (Zn-doped oxides), OER kinetics is limited by the formation of oxo groups [
200].
Figure 13. Decrease in the binding energy from Zn- to Mn-doped nickel oxides showed a volcano-type OER activity by changing the rate-determining step from •O formation to O-O coupling. Reproduced from
J. Am. Chem. Soc. 2022 [
200] with permission from the American Chemical Society.
Oxygen vacancy-enriched porous NiO/ln
2O
3 nanofibers (Vo–NiO/ln
2O
3@NFs) were fabricated for efficient OER electrocatalysis. Abundant Vo modulated the electronic configuration of the catalyst for altering the adsorption of intermediates to reduce the OER overpotential and promote *O formation, upshifting the d band center of metal centers near the Fermi level, and also increasing the electrical conductivity and enhancing the OER reaction kinetics simultaneously. In situ Raman spectra suggested that the Vo can render the NiO/ln
2O
3 more easily reconstructible on the surface during the OER course [
201].
DFT +U calculations revealed that Ir-doping of a β-NiOOH(001) surface enhanced the electric conductivity while also activating an oxygen site involving three Ni atoms to realize a remarkably low OER overpotential of only η = 0.46 V, much lower than the oxygen site involving three Ni atoms in pristine β-NiOOH (η = 0.66 V) [
202]. Since theoretical calculations predicted that Co, Rh, and Ir dopants would lead to low overpotentials to improve the OER activity of Ni-based hydroxides, an experimental confirmation on the altered OER activities for a series of metals (Mo, W, Fe, Ru, Co, Rh, Ir) doped into γ-NiOOH has been reported [
203]. The in situ electrical conductivity for metal-doped γ-NiOOH correlated well with the trend in enhanced OER activities. The DFT calculations, which suggested that the intrinsic connections to the double exchange interaction between adjacent metal ions with various d orbital occupancies, rationalized the experimental results, serving as an indicator for the key metal-oxo radical character [
203].
7.3. FeOx
Recent advancement and progress in initializing Fe-based OER electrocatalysts with different supporting materials, including carbon-based materials, layered double hydroxides, Prussian blue analogous, metal–organic frameworks, were reviewed by Xiong et al. [
204]. In the review, the OER mechanism and some typical OER electrochemical parameters of Fe-based electrocatalysts supported on various supporting materials from experimental and theoretical viewpoints were highlighted. Some challenges and expectations for promoting the catalytic performance were described [
204].
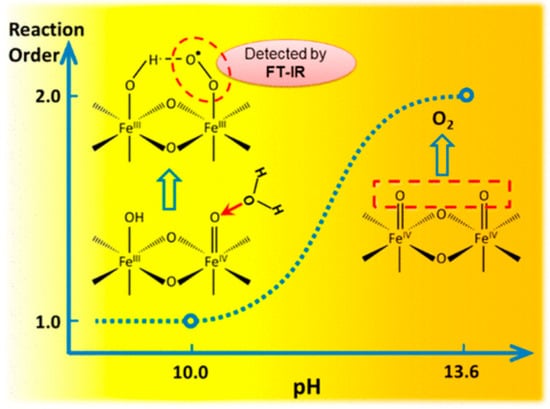
In photoelectrochemical (PEC) water oxidation on hematite (α-Fe
2O
3), the mechanism of the subsequent rate-limiting O–O bond formation step was investigated by rate law analysis based on EIS measurements and probing the reaction intermediates with operando FTIR spectroscopy. Distinct reaction orders of ~1 and ~2 were observed in near-neutral and highly alkaline environments, respectively. The unity rate law in near-neutral pH regions suggests a mechanism of WNA to –Fe=O to form the O–O bond. Operando observation of a surface superoxide species by FTIR further confirmed this pathway. In highly alkaline regions, coupling of adjacent surface trapped holes (I2M) becomes the dominant mechanism. While both are operable at intermediate pHs, the mechanism switch from I2M to WNA induced by local pH decrease was observed at a high photocurrent level as shown in
Figure 14 [
87]. In the recent report, transient photocurrent measurements for hematite photoanodes revealed that the OER rate has a third-order dependence on the surface hole density. A mechanism wherein the reaction proceeds by accumulating oxidizing equivalents through a sequence of one-electron oxidations of surface hydroxy groups was proposed. The key O–O bond formation step occurs by the dissociative chemisorption of a hydroxide ion involving three oxyl sites [
205].
Figure 14. In photoelectrochemical water oxidation on α-Fe
2O
3, the OER mechanism switches from WNA to I2M at a strong alkaline solution. Reproduced from
J. Am. Chem. Soc. 2018 [
87] with permission from the American Chemical Society.
Polycrystalline γ-FeO(OH), synthesized at room temperature, was used as a stable, although reactive, anode for OER, and electrokinetic studies were performed to unravel the OER pathway [
206]. The cell temperature, hydroxyl ion concentration, and the cation of the supporting electrolyte were varied, and the influence of external bias on the OER activity was recorded. Tafel slope and charge-transfer resistance values at high temperatures up to 65 °C unambiguously highlight the influence of the thermodynamic barrier and electron transfer kinetics. The faster OER kinetics on polycrystalline γ-FeO(OH) can also be attributed to an appreciably low activation energy, where the variation of the electrolyte concentration indicated a first-order dependence on the OH
− concentration. The deuterium isotope effect implicated the dissociation of hydroxyl ions on the polycrystalline γ-FeO(OH) as the rate-determining step. The direct effect of cations such as Li, Na, and K of the electrolyte on OER indicated a weak interaction of the cations with the surface-active [Fe
III-OH] species [
206].
Fe
3O
4 with oxygen vacancies (Fe
3O
4-V
O) was synthesized via the Ar ion irradiation method and its OER activity was greatly improved by properly modulating the electron density around Fe atoms, which were evaluated with XANES and EXAFS methods. DFT results indicated the enhancement in desorption of the *OOH groups, which significantly reduced the OER reaction barrier. Actually, the Fe
3O
4-Vo catalyst showed a better overpotential than commercial RuO
2 [
207].
Ni-, Co-, and Yb-doped FeO(OH) nanorod arrays grown directly on carbon cloth (CC) are synthesized by a simple one-step hydrothermal method. The doped Ni
2+ and Co
2+ can occupy Fe
2+ and Fe
3+ sites in FeO(OH), increasing the concentration of oxygen vacancies and the doped Yb
3+ with a larger ionic radius can occupy the interstitial sites, which leads to more edge dislocations. The oxygen vacancies and edge dislocations greatly enrich the active sites in FeO(OH)/CC. In addition, DFT calculations confirmed that doping Ni
2+, Co
2+, and Yb
3+ modulates the electronic structure of the main active Fe sites, bringing its d-band center closer to the Fermi level and reducing the Gibbs free energy change in the rate-determining step of the OER [
208].
7.4. MnOx
Nature uses a Mn cluster for water oxidation in PS II, and thus, water oxidation using Mn clusters is interesting in artificial water-splitting systems. An ultra-thin manganese oxide (MnO
X) was selected as a co-catalyst to modify the surface of BiVO
4 photoanode by a spray pyrolysis method [
209]. The PEC measurements demonstrated that the surface charge transport efficiency strikingly increased by the MnO
X modification. After applying Ar plasma to the BiVO
4/MnOx sample, the transport efficiency further increased, and it was around 7 times higher compared with that of pristine BiVO
4 samples. The remarkable PEC performance could be attributed to the increased charge carrier density, extended carrier lifetime, and additional exposed Mn active sites on the BiVO
4 surface [
209].
An α-Mn
2O
3/FTO electrocatalyst was used in nonaqueous (CH
3CN and DMF) and aqueous 0.1 M KPi (pH 7.0) solutions for kinetic studies of heterogeneous water oxidation. The rate of water oxidation was first order in catalyst concentration and in H
2O concentration. The square wave and cyclic voltammetry measurements revealed the stepwise proton-coupled electron transfer oxidations of the active Mn
II–OH
2 site to Mn
III–OH and then to Mn
IV=O and finally an electron transfer oxidation of Mn
IV=O to Mn
V=O species. The Mn
V=O species undergoes a rate-limiting O atom transfer to H
2O to give a Mn
III–OOH
2 species that, in turn, undergoes further oxidations to release O
2 [
61].
An Mn–K cluster was investigated for electrochemical water oxidation. By using XAS, SEM, TEM, XRD, FTIR spectroscopy, and electrochemical methods, it was revealed that conversion into nanosized Mn oxides occurred for the cluster, and the nanosized Mn oxides are the true catalyst for water oxidation [
210].
The Mn
3O
4 nanocatalyst, which exhibits superb catalytic activity for water oxidation under neutral conditions, was analyzed for the complex capacitance. By the change in Mn valence between Mn
II and Mn
IV, the charge was accumulated on the catalyst surface prior to the rate-determining O–O bond-forming step. The dissipation ratio was proposed for understanding the energy balance between charge accumulation and charge consumption for chemical O–O bond formation [
211]. In Mn
3O
4 nanoparticles, a profile imaging technique was exploited to understand the correlation between surface atomic structures and the OER. The surface structures of Mn
3O
4 nanoparticles were changed by the reaction and the surface Mn ions were reconstructed. The commonly considered active sites disappeared from the reconstructed planes, whereas Mn ions were still exposed at the edges of nanoparticles. Thus, the surface reconstructions can deactivate low-index surfaces of Mn oxides in the OER process, which was further validated by DFT calculations [
212].
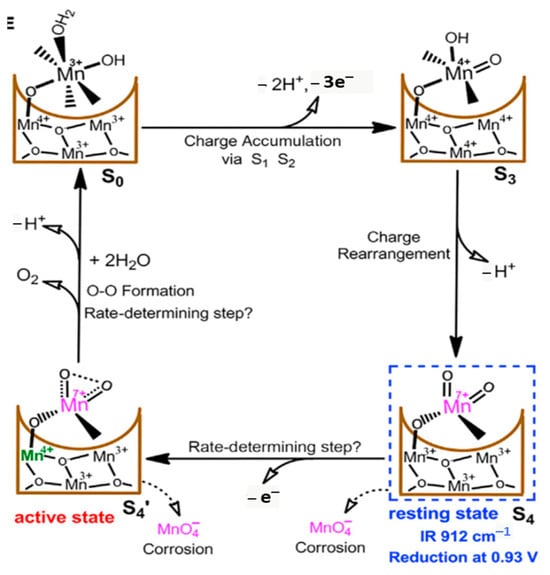
An Mn
VII=O intermediate during electrocatalytic water oxidation by a c-disordered δ-MnOx was identified as an onset-potential-dependent reduction peak at 0.93 V. This intermediate is proven to be highly reactive and much more oxidative than permanganate ion. Thus, a new catalytic mechanism for water oxidation catalyzed by Mn oxides was proposed with the involvement of the Mn
VII=O intermediate in a resting state and the Mn
IV-O-Mn
VII=O as a real active species for O-O bond formation.
Figure 15 shows the proposed catalytic cycle, involving Mn
VII=O, in the MnOx-catalyzed water-oxidation reaction. The overall mechanistic process involves charge accumulation (S
0/S
3), charge rearrangement (S
3/S
4), active-state formation (S
4/S
4′), and oxygen evolution (S
4′/S
0) [
68].
Figure 15. Proposed mechanism in MnO
x-catalyzed OER. Reproduced from
iScience 2018 [
68] under the license of CC BY 4.0 of Elsevier.
7.5. Mixed Metal Oxides
Two or three transition metals are mixed to form oxides of high electrocatalytic performance for water electrolyzers at a low cost. An NiFe oxide catalyst was employed as the anode catalyst with an NiMo oxide cathode catalyst with a high-performance perovskite-Si tandem solar cell, achieving a record 20% STH efficiency [
213]. Nickel ferrite, NiFe
2O
4, cobalt ferrite, and CoFe
2O
4, are efficient and promising anode catalyst materials in the field of electrochemical water splitting.
In Ni-Fe water oxidation electrocatalysts, Ni is likely not an active site for water oxidation, because Ni cannot achieve high-oxide states in aqueous environments at relevant potentials [
214]. For the OER of NiFeO
xH
y, the addition of Co
2+ cation increased the current density by 32.7% by the cation transport effect [
215]. Using operando XAS, it was revealed that Ni oxidizes from the initial +2 oxidation state to the +3/+4 state [
216]. For Ni-Fe oxyhydroxides, in situ monitoring of the Fe active site number and turn-frequency number provided important insights into the activity degradation/regeneration caused by Fe dissolution/adsorption as well as site-dependent activity and stability [
217]. In the case of NiFe
2O
4, an Fe-site-assisted LOM pathway as the preferred OER mechanism was predicted. On the other hand, in the case of CoFe
2O
4, an Fe-site-assisted LOM pathway and a Co-site-assisted AEM pathway could both play a role [
218].
Amorphous/crystalline NiFe
2O
4 induced by vanadium doping showed a superior electrocatalyst and long-term stability [
219]. For amorphous Ni-Fe mixed metal oxides, analysis of the XAS revealed local structural transitions. A dual-site OER reaction mechanism was proposed, in which potential and rate-determining steps occur at Ni and Fe sites, respectively [
220].
An Fe/Ca-based bimetallic oxide, CaFe
2O
4, exhibited outstanding OER activity in alkaline media. DFT calculations suggested an unconventional mechanism via the direct formation of O–O bonds between two oxygen intermediates, which are adsorbed on a multi-iron site on the catalyst surface [
221].
On spinel NiCo
2O
4 abundant Co defects were preferentially produced by tuning the M–O bond length. Theoretical calculations and experiments proved that Al doping elongated the Co–O bond and promoted the ionization of Co under plasma treatment [
222]. Spinel Co
2MnO
4 showed higher OER activity, most probably due to the ideal binding energies of the OER intermediates [
223].
For modulated NiFeX and FeCoX (X = W, Mo, Nb, Ta, Re, and MoW) oxyhydroxide catalysts, in situ and ex situ soft and hard XAS were used to characterize the oxidation transition and facilitate the lower OER overpotential [
224]. (Co–Fe–Pb)Ox in acidic solutions through a cobalt-selective self-healing mechanism was investigated. The kinetics of the process were investigated by soft XAS and it was revealed that low concentrations of Co
2+ in the solution stabilize the catalytically active Co(Fe) sites [
225].
8. Layered Double Hydroxide (LDH)
LDH are emerging catalyst materials with inner layer water molecules and higher anion exchange capacity. They have been extensively used as electrocatalytic materials owing to their high specific surface area, environmental friendliness, lower cost, and non-toxicity [
226]. A kind of LDH itself may become photocatalysts for water splitting. The electronic properties, such as band structure, bandgap energy (Eg), density of states (DOS), and band edge placement for M
IIM
III-LDHs (MI
II = Mg, Co, Ni and Zn; M
III = Al and Ga) were calculated by using the DFT + U method. The band structures of Mg- and Zn-based LDHs and Co- and Ni-based LDHs are responsive to ultraviolet (Eg > 3.1 eV) and visible light (Eg < 3.1 eV), respectively. The DOS calculations revealed that the photogenerated hole localizes on the surface hydroxyl group of LDHs, facilitating the oxidization of a water molecule without a long transportation route. The band edge placements of NiGa-, CoAl-, ZnAl-, and NiAl-LDHs have a driving force (0.965 eV, 0.836 eV, 0.667 eV, and 0.426 eV, respectively), toward oxygen evolution. In the experimental observations, only CoAl-LDH was an efficient oxygen evolution photocatalyst, agreeing well with the theoretical prediction [
227].
For NiFe-LDH and Ni-LDH, the critical role of superficial oxygen vacancies in enhancing electronic transport was discussed based on the electrochemical analysis by correlating with electrocatalytic activities [
228]. The in situ conversion process to yield a monolayer of Ni(OH)
2 on electrodes was presented and the dynamic active site of the monolayer promoted the OER process. Doping with Co caused the oscillation of Ni and Co valence states in NiCo hydroxide. This study defined an in situ conversion process to yield monolayer LDH and a fundamental understanding of the origin of the active sites in monolayer LDHs for the OER [
229]. Direct spectroscopic evidence for the different active sites in Fe-free and Fe-containing Ni oxides was reported for ultrathin LDH samples.
18O-labeling experiments in combination with in situ Raman spectroscopy were employed to probe the role of lattice oxygen as well as an active oxygen species, NiOO
−, in the catalysts. It was found that lattice oxygen is involved in the OER for Ni and NiCo-LDHs, but not for NiFe and NiCoFe-LDHs. Moreover, NiOO
− is a precursor to oxygen for Ni and NiCo-LDHs, but not for NiFe and NiCoFe-LDHs [
78]. For M-doped Ni-based LDH (M = Ni, Co, and Fe), the OER mechanism was investigated theoretically for the reaction processes of AEM, LOM, and IMOC (= intramolecular oxygen coupling) mechanism. Theory predicted overpotential and Tafel slopes, and the findings were in agreement with the observation. As a result, depending on the applied potential, the reaction mechanism changed [
230]. In addition to electrocatalysts, NiFe-LDH may be used as the flexible electrode of Zinc-Air batteries [
231].
For the Cu-NiFe-LDH electrocatalyst, a novel magnetic Fe
III site spin-splitting strategy was suggested [
232]. The electronic structure and spin states of the Fe
IIII sites are effectively induced by the Jahn–Teller effect of Cu
2+. The theoretical calculations and operando ATR FTIR revealed that the facilitation for the O–O bond formation accelerated the production of O from OH and improved the OER activity [
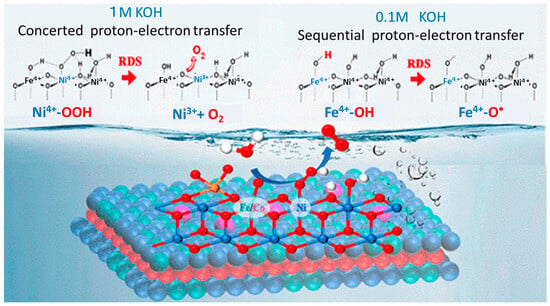
232]. For as-prepared sulfated Co-NiFe-LDH nanosheets, the kinetic energy barrier of the O–O coupling is significantly reduced. The formation of M-OOH on the active site at low overpotential was directly confirmed in 1 M KOH solution by in situ Raman and charge transfer fitting results. In a weakly alkaline environment of 0.1 M KOH, a sequential proton–electron transfer mechanism replaces the concerted proton–electron transfer mechanism, and the proton transfer step becomes the rate-determining step (RDS) as illustrated in
Figure 16 [
233].
Figure 16. For Co-NiFe-LDH nanosheet, weakening alkaline concentration changes rate-determining step (RDS) from the release of O
2 to the oxo radical formation. Reproduced from
ACS Nano 2023 [
233] with permission from the American Chemical Society.
A Pt-induced NiFe-LDH (Pt-NiFe LDH) nanosheet was synthesized and large current density electrodes could be achieved in OER as well as HER [
234]. At NiO/NiFe-LDH, the adsorption energy of *OH and *OOH can be adjusted independently, so as to bypass the scaling relationship and achieve high catalytic performance [
235]. ZnO nanoparticles are uniformly distributed on the NiFe-LDH nanoflowers, which are prepared uniformly on the three-dimensional porous Ni foam. The active sites changed from Fe cations to Ni cations during OER and the OER dynamics were significantly improved [
236]. The hierarchical bimetal nitride/hydroxide (NiMoN/NiFe LDH) array exhibits the industrially required current density. In situ electrochemical spectroscopy reveals that a hetero interface facilitates dynamic structure evolution to optimize the electronic structure. Operando EIS measurement implied the accelerated OER kinetics and intermediate evolution due to fast charge transport. For the OER mechanism, the combination of theoretical and experimental studies revealed that as-activated NiMoN/NiFe-LDH follows an LOM process with accelerated kinetics [
237].
NiFe LDH@Ni
3S
2 heterostructure as an efficient bifunctional electrocatalyst for overall water splitting was prepared. Three-dimensional porous heterostructure arrays caused good electrocatalytic activity with a low Tafel slope [
238]. By incorporating a semiconductor CdS/CdSe-MoS
2 and NiFe-LDH for the OER, the as-prepared photoelectrode required a potential lower than the theoretical water splitting potential. Operando XAS measurements revealed that the formation of highly oxidized Ni species under illumination provides large photocurrent gains [
239]. Hetero structures of LDH with graphitic carbon nitride (g-C
3N
4) stand as promising photo- and electro-catalysts for water oxidation and reduction. Mechanisms involved in electrocatalytic, photocatalytic, and photoelectrocatalytic water splitting processes were reviewed with the necessary insights on the material [
240]. By taking CuTi-LDH@g-C
3N
4 and Bi
2O
2CO
3/NiFe-LDH@g-C
3N
4 as examples, the importance of heterojunctions and interfacial chemistry in the water-splitting mechanism was explained in detail [
240].