1. DC-Based Anticancer Vaccines “101”
The application of vaccines in oncology has generated considerable excitement in recent years and represents a potential paradigm shift in how cancer could be treated and ultimately prevented. Three types of vaccine platforms or formulations have been developed for cancer therapy: cellular vaccines, vector-based vaccines, and molecular vaccines (DNA, RNA, peptides, and proteins) [
37,
38]. Despite the recent focus on molecular vaccines, the DC vaccine strategy (which falls under the cellular vaccine category) represents an exciting therapeutic avenue. With this strategy (shown in
Figure 1), DCs’ superior antigen presentation machinery is exploited, whereby DCs are tumour-associated antigen (TAAs)-loaded and matured ex vivo before their autologous reinfusion into cancer patients in order to bypass TME immunosuppressive mechanisms and generate TAA-specific CTLs to initiate tumour cell killing and induce long-term immunological memory [
2]. Although various clinical trials have confirmed the safety and feasibility of DC vaccines to induce an anti-tumour response, clinical patient responses remain poor [
39]. Sporadic clinical responses are unsurprising considering the lack of a standardized protocol and the numerous variable factors involved in the design and execution of a DC vaccine strategy [
40]. Such variable factors include the source and production of a DC cell type with potential inherent suboptimal antigen presentation and migration capacity. DC maturation stimuli, route of vaccine administration, frequency of injection, additional adjuvants, and overall competence of the patient’s immune system can also influence clinical outcomes. Initial DC vaccine strategies were designed to target advanced cancers, meaning the well-established active immunosuppression mechanisms of the TME provided a significant barrier to efficacy. The genomic instability of late-stage cancers results in heterogeneous tumour antigen expression, which again can hamper vaccines designed around a single epitope-targeting strategy. Despite these obstacles, the main technical issue with DC vaccines that needed to be overcome in order to make it a feasible translational avenue was the limited source material due to the low prevalence of DCs in the peripheral blood, ranging from 0.1–1% of peripheral blood mononuclear cells (PBMC) [
41]. The original vaccine preparation attempts used density gradients to isolate an analogous APC-enriched preparation (leukapheresis) from a cancer patient’s own blood. This early strategy yielded Sipuleucel-T (Provenge
®; Dendron Corporation, Seattle, WA, USA), a DC vaccine against asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer [
42]. Sipuleucel-T was generated by exposing ex vivo cultured analogous APC-enriched preparations to a recombinant fusion protein consisting of the tumour antigen prostatic acid phosphatase (PAP) and granulocyte-macrophage colony-stimulating factor (GM-CSF). The IMPACT trial subsequently confirmed that Sipuleucel-T increased median overall survival (OS) rates by 3.9 months for castration-resistant prostate cancer patients [
42]. Although modest, this clinical outcome was crucial in demonstrating the safety of the DC vaccine approach and led to subsequent FDA approval in 2010. Currently, further trials are underway with Sipuleucel-T in combination with other anticancer therapies in order to increase response rates [
43].
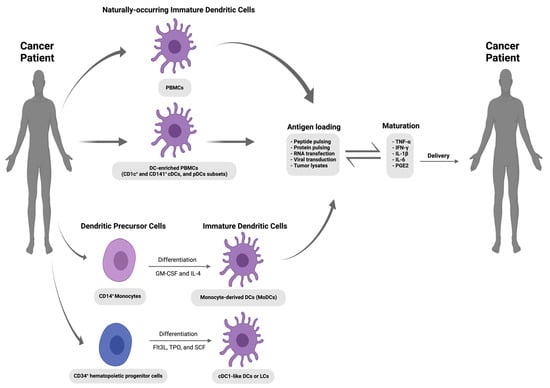
Figure 1. Schematic representation of the DC-based anti-cancer vaccination strategy. Immature dendritic cells are isolated from blood (peripheral blood mononuclear cells [PBMCs] or DC-enriched PBMCs) or derived from blood monocytes (MoDCs) and CD34
+ hematopoietic stem progenitor cells (HSPCs). After ex vivo activation and antigen loading, these autologous DCs could be re-administered into the cancer patient via various routes of administration (I.V., intravenous; I.N., intranodal; I.D., intradermal; and S.C., subcutaneous) to induce an antigen-specific T cell response against tumours [
44]. The figure was created using
www.app.biorender.com (accessed on 12 October 2023).
2. Genetic Engineering of DC-Based Vaccines to Improve Their Immunotherapeutic Potentials—A from within Approach
2.1. Viral-Based Approaches
Due to viruses’ natural capability to efficiently transduce eukaryotic cells with foreign nucleic acids, viral vectors have consistently been utilised as an efficient delivery vehicle for genetic modification purposes in academic and industrial laboratories for both research and clinical gene therapy applications. Viral vectors are classified according to whether viral infection results in transient short-term gene expression or permanent long-term gene expression following integration into the host genome. Additionally, depending on their genetic makeup, viruses can be classified into RNA-based and DNA-based vectors with either single-stranded (ss) or double-stranded (ds) genomes. Examples of the viral vectors include γ retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses [
61].
2.1.1. CCR7
Manipulation of DCs’ ability to migrate to lymphoid tissues in order to prime CD8
+ CTLs could dramatically improve DC vaccine efficacy. Numerous animal models have established that the chemokine receptor C-C chemokine receptor type 7 (CCR7), a G-protein-coupled receptor for the chemokine ligands CCL19 and CCL21, regulates DC chemotaxis, survival, and migratory speed in lymphoid tissue [
62]. Indeed, high CCR7 expression levels in human tumours correlate with better clinical outcomes [
63]. In 2005, Okada et al. proposed that genetically engineered DC vaccines to enhance CCR7 expression could produce a TAA-loaded DC that would efficiently home to a nearby lymphoid tissue and activate a robust T-cell response after administration—a strategy called “lymphoid tissue-directivity DC vaccine” [
58]. Hence, a CCR7-overexpression bone marrow-derived DC vaccine was created by viral gene transduction using replication-deficient AdRGD (an adenovirus serotype 5 backbone with deletions of the E1 and E3 regions and the RGD sequence for AV-integrin targeting). Subsequent efficient AdRGD-mediated CCR7-gene transduction and overexpression into BMDCs were confirmed by semi-quantitative qRT-PCR analysis. Additionally, they reported that BMDCs—overexpressing CCR7 and cultured for 24 h—exhibited sufficient CCR7 protein localization on the cell surface in the flow cytometric analyses and showed strong migratory ability toward a CCL21 concentration gradient in a chemotaxis assay. Moreover, BMDCs expressing enhanced green fluorescent protein (eGFP) and transduced with AdRGD-CCR7 were demonstrated to migrate into the regional lymph nodes at approximately a 15-fold higher rate compared with mock DCs upon intradermal injection into the flank of wild-type mice.
2.1.2. CD40L
CD40 Ligand (CD40L) or (CD154) is a type II transmembrane protein and a member of the tumour necrosis factor (TNF) superfamily of protein ligands [
64]. Stimulation of the CD40 receptor on DCs by CD40L on activated CD4
+ T lymphocytes (a process called DC licensing) results in the upregulation of co-stimulatory molecules (CD80/CD86) on DC surfaces, promotes proinflammatory cytokine production (e.g., IL-12, TNF-α, and interferon-γ (IFN-γ), facilitates the cross-presentation of antigens [
64,
65], and up-regulates CCR7 expression to enhance the capacity of DCs to migrate into secondary lymphoid tissues [
66]. IL-12 is considered a Th1-polarising cytokine in both mouse and human DCs [
4]. Additionally, it delivers a crucial cue for both Th1 T cell and CD8
+ T cell differentiation and functions to induce potent anti-tumour cytotoxic T-cell immune responses. Ex vivo maturation of MoDCs by incubation with the Jonuleit cytokine cocktail showed some drawbacks [
67,
68]. For instance, matured MoDCs were found to be able to migrate but lack IL-12p70 expression [
69]. To overcome this drawback, Ilka Knippertz and colleagues (2009) transduced MoDCs with an adenovirus vector coding for the trimeric human CD40L (Ad5hCD40L) in the presence of the Jonuleit cytokine cocktail in combination with a recombinant human IFN-γ treatment, which resulted in an increase in IL-12p70 expression and migration towards CCL19 [
70].
2.2. RNA-Based Approaches
2.2.1. CD40L and TLR4
CD40L was introduced in the previous section and can increase DC expression of co-stimulatory molecules and ultimate maturation. Toll-like receptor 4 (TLR4) is a pattern recognition receptor that plays a central role in initiating innate immune cell responses and is involved in DC activation and pro-inflammatory cytokine production [
71]. Hence, overexpressing TLR4 in DCs was determined to be a novel route to activate DCs without the need for cytokine cocktails. An example of a strategy to deliver mRNA in DCs was described by Calderhead et al. (2008) [
72].CD40L mRNA was delivered by electroporation to cytokine-matured (treated with IL-1β, TNFα, IL-6, and PGE2) MoDCs, resulting in the long-term expansion of MART-1-reactive T cells that showed a CD28
+/CD45RA
− effector/memory phenotype. In terms of TLR4 manipulation, Cisco et al. (2004) [
73] showed that electroporation of a mRNA encoding a constitutively activated version of TLR4 (caTLR4) RNA into human DCs could lead to significant cytokine production and DC maturation (without the need for IL-12/TNF-γ-specific maturation) that led to enhanced allostimulation of CD4
+ T-cells. Bonehill et al. (2008) combined both approaches through the delivery of mRNA species for CD40L, caTLR4, and CD70 into immature MoDCs as mRNA [
74]. They demonstrated in vitro that the specific introduction of CD40L and caTLR4 into immature MoDCs could generate mature, cytokine-secreting DCs, mimicking CD40-CD40L and TLR4-LPS ligations. Furthermore, the co-introduction of CD70 provided a co-stimulatory signal to CD27
+ naïve T-cells capable of blocking activated T-cell apoptosis and supporting T-cell proliferation. This combination was named TriMix and has been taken forward into preclinical trials where, instead of antigen pulsing, TriMix and MelanA-encoding (tumour-associated antigen) mRNA were electroporated together into DCs [
35]. Results demonstrated the ability of this strategy to activate CD8
+ T cells specific for tumour antigens. In a recent phase II clinical trial, TriMixDC-Mel in combination with ipilimumab was used to treat patients with pre-treated advanced melanoma [
75]. In total, 39 patients were treated, and primary end-point data were collected at six months, where 38% of patients demonstrated anti-tumour responses. There were some common adverse events, such as skin reactions at the injection site (100%) and flu-like symptoms (84%); however, there were no grade 5 adverse events, and grade 3 or 4 immune-related adverse events were 36%. It was therefore concluded that this treatment was tolerable and showed tumour responses in the patient cohort tested. Fifteen patients were followed up for 5+ years, and their immune stimulation was measured [
76]. After 390 weeks, 11 patients were alive, with 7 in complete remission. CD8
+ T-cell responses for tumour-associated antigens were seen in all patients. Such findings clearly highlight the considerable potential for DC vaccines in vivo.
2.2.2. IKKα and IKKβ
Ubiquitous NFκB transcription factors are central coordinators of immunity, inflammation, and cell survival [
77]. In mammals, NFκB comprises a family of five proteins, forming ubiquitous dimeric complexes, NFκB1(p50)/RelA(p65) being the most abundant. In the canonical NFκB pathway, these dimers are normally held inactive, bound to IκB-family inhibitory proteins, and can be activated by signals causing the phosphorylation and proteolysis of IκBs by the IκBα-kinase (IKK) complex (typically comprising the two homologous catalytic subunits, IκB kinase α (IKKα) and IκB kinase β (IKKβ) and the regulatory scaffold subunit NEMO) and the proteasome, respectively [
78]. NFκB thereafter enters the nucleus to activate transcription of a multitude of inflammatory mediators, immunoregulators, apoptosis inhibitors, and other genes, moulding the host defence responses to stress, injury, and infection [
77]. NFκB pathway activation has been demonstrated to be crucial for DC maturation [
79,
80], and consequently, the overexpression of certain components of this pathway, in particular IKKα and IKKβ, could potentially open an additional avenue for mature DCs. Therefore, Pfeiffer et al. (2014) thought to activate this pathway through electroporation of constitutively active mutants of IκB kinases (caIKKα and caIKKβ) mRNAs in cytokine-matured human MoDCs [
81]. DCs expressing these kinases had upregulated maturation markers, secreted higher cytokine levels (including IL-12), and induced CD27 expression in T cells in vitro. CD27 expression on T-cells indicates a memory phenotype demonstrating enhanced expansion capability, unlike traditional cytokine-maturated DCs alone. A follow-up study also demonstrated that these DCs can robustly activate autologous NK cells (which can directly lyse and kill tumour cells), as shown by the upregulation of CD54, CD69, and CD25 in vitro [
82].
2.2.3. PD-L1 and PD-L2
Programmed death 1 (PD-1) is involved in the control of immune tolerance and is one of the main protagonists in immune escape mechanisms during chronic viral infections and cancer [
83,
84]. The ligands of PD-1 (PD-L1 and PD-L2) are type I transmembrane glycoproteins that, upon binding to PD-1 on activated T and B cells, block T cell proliferation, cytokine production, and cell adhesion [
83,
84]. PD-L1 and PD-L2 expression have been demonstrated on the surface of APCS, including DCs. Consequently, siRNA silencing of PD-L1 and PD-L2 was undertaken by Hobo et al. (2010) in MoDCs [
60]. Compared with the non-electroporated controls, these cells were shown to have the capacity to efficiently expand CD8
+ effector and memory T cells in leukemic patients. Overall, DCs with PD-L1 and PD-L2 knockdown were demonstrated to enhance T cell proliferation and cytokine production. More recently, instead of electroporation, Hobo et al. showed that they could deliver the PD-L1 siRNA in lipid nanoparticles, resulting in the same DC phenotype as above [
59].
2.2.4. PTEN
Phosphatase and tensin homologue (PTEN) is a key negative regulator of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which is involved in the activation of DCs [
85,
86]. Therefore, Kim et al. (2010) hypothesized that inhibition of PTEN could lead to heightened activation of DCs through unlocking the PI3K/AKT axis [
87]. Indeed, down-regulation of PTEN in bone marrow-derived DCs (BMDCs) by transfection of PTEN siRNA was shown to result in AKT-dependent DC maturation [
54]. Also observed was the upregulation of co-stimulatory molecules and the chemokine receptor CCR7, which demonstrated higher levels of in vitro T cell activation and generated higher numbers of tumour-specific CD8
+ T cells in the HPV-16 E7-expressing murine tumour model [
54].
2.3. CRISPR/Cas9-Based Engineering of DCs
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system was discovered as a part of the adaptive immune system in bacteria that introduces site-specific double-stranded breaks (DSBs) in target foreign DNA by means of the dual RNA-guided DNA endonuclease Cas9 [
88]. Since then, it has been widely used to modify targeted genes in eukaryotic cells and organisms and has emerged as a powerful tool for genome engineering. The induced DNA DSBs are usually repaired in eukaryotic systems by specific DNA-repair pathways (homology-directed repair [HDR], microhomology-mediated, end joining [MMEJ], and non-homologous end-joining (NHEJ]) [
89]. Homology-directed repair (HDR) is a precise repair mechanism that depends on a homologous DNA template (a homologous sequence flanking the DNA cut site) to guide the repair of DSBs. MMEJ leads to deletions of DNA stretches of various lengths at the DNA break sites, resulting in the loss of sequence information. NHEJ leads to random insertions or deletions (InDels) of base pairs, resulting in frame shift mutations. Accordingly, MMEJ and NHEJ repair pathways functionally inactivate targeted genes with high efficiency [
88]. The recent advancements of CRISPR/Cas9 gene-editing methodologies have made them more efficient (high specificity with minimal off-target effects), technically feasible, and promising to be included in DC-based vaccine manufacturing. If the actionable target genes that are implicated in the dysfunctional anti-tumoral roles of DCs do not severely affect the cell-cycling or cell viability phenotypes, such a strategy could be valuable in creating more potent genetically manipulated DC-based vaccines. Recent investigations, outlined below in
Figure 2, revealed adverse molecular roles of several specific DC genes in the milieu of the tumour microenvironment (TME). It is worth noting that many other genes and signalling pathways have been suggested to be targeted to enhance the DC activation process. For instance, in the presence of TGF-β [
90], DCs acquire a regulatory phenotype that favours the promotion of a tolerogenic immune response—a rationale to target TGFBR1 and TGFBR2 for genetic manipulation in DC-based vaccines. Additionally, interleukin-10 (IL-10) [
91] produced by DCs efficiently inhibits proliferative and cytokine responses in T-cells, mediating immunological unresponsiveness and suppression of immune reactions—another rationale to ablate IL-10 in DC-based vaccines. However, the genes outlined below were reviewed based on favourable anti-tumoral phenotypes achieved by in vitro and/or in vivo knockdown or knockout in the context of tumorigenesis. Such experimental interventions have been demonstrated to be effective at enhancing anti-tumoral DC function in pre-clinical models.
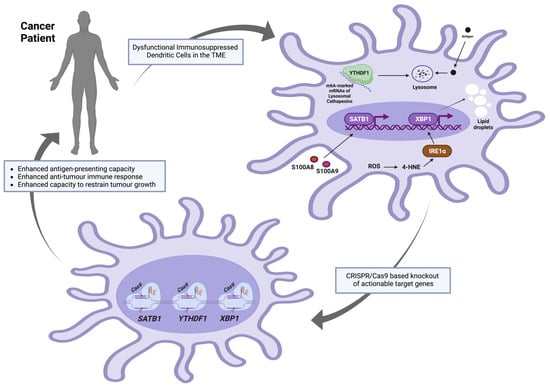
Figure 2. Proposed target genes to be knocked out in DCs using CRISPR/Cas9 approaches prior to autologous transfer to cancer patients. The figure was created using
www.app.biorender.com (accessed on 12 October 2023).
2.3.1. YTHDF1
YTH N6-methyladenosine RNA binding protein F1 (YTHDF1) is a member of a protein family—called “readers”—that can recognise the N6-methyladenosine (m6A) methylation as a post-transcriptional modification of mRNA transcripts. This family contains the YT521-B homology (YTH) domain; hence, the name YTHDF protein family [
95]. In dendritic cells, YTHDF1 could bind to m6A-marked mRNAs encoding lysosomal cathepsins (proteases responsible for lysosomal-specific antigen degradation), promoting their translation and negatively affecting antigen presentation capabilities [
57]. Recently, Dali Han and colleagues showed that the extent to which Ythdf1
−/− FLT3L-DCs (conventional DCs pulsed with necrotic B16-OVA cells in vitro) could cross-prime T cells expressing transgenic ovalbumin-specific (OT-I) T cell receptors was higher than the wildtype DCs. Additionally, at an in vivo level, CD8α
+ and CD11b
+ DCs from draining lymph nodes (DLNs) of B16-OVA- or MC38-OTIp-tumour-bearing Ythdf1
−/− mice showed a substantially augmented cross-priming capacity as compared with those collected from the wild mice when both co-cultured with OT-I T cells. Such an enhanced cross-priming capacity of CD8
+ T-cells was attributed to the improved antigen presentation by DCs (as assessed by the abundance of H-2Kb-SIINFEKL complexes on DCs from wild-type and Ythdf1
−/− mice bearing B16-OVA tumours).
2.3.2. XBP1
X-box binding protein 1 (XBP1) is a major transcription factor and a downstream effector of the inositol-requiring enzyme 1 (IRE1α) in the unfolded protein response (UPR)—a process that is usually initiated by endoplasmic reticulum (ER) membrane-bound sensors [
96] to ensure protein folding fidelity and relieve the load of unfolded or misfolded proteins, restoring protein homeostasis (proteostasis) [
97]. IRE1α-XBP1 activation and overexpression of various endoplasmic reticulum (ER) stress response markers were reported in ovarian cancer-associated DCs as compared with DCs isolated from non-tumorigenic normal tissues. This DC-specific ER stress was a result of the elevated levels of intracellular reactive oxygen species (ROS) that induced lipid peroxidation-generating reactive by-products (for example, the unsaturated aldehyde 4-hydroxy-trans-2-nonenal (4-HNE)) that could alter the ER-resident chaperone functions. Compounds that could sequester ROS or lipid peroxidation by-products (vitamin E or hydralazine, respectively) were reported to inhibit Xbp1 mRNA splicing and prevent the induction of the expression of ER stress response genes in ovarian cancer-associated DCs. Most importantly, the development and progression of p53/K-Ras-driven primary ovarian cancer were compromised in irradiated mice that were reconstituted with XBP1
f/f CD11c-Cre donor bone marrow as compared with control hosts transplanted with XBP1-sufficient (XBP1
f/f) littermate bone marrow. Additionally, the same effects were observed in metastatic ovarian cancer models and were accompanied by the infiltration and accumulation of CD44
+ IFNγ-secreting CD8
+ and CD4
+ T cells at tumour beds. Accordingly, the expression of XBP1 in CD11c
+ DCs was reported to be critical for the initiation and progression of ovarian cancer, and its conditional ablation in DCs in the tumour microenvironment enhanced the capacity to restrict tumour growth. Finally, it was reported that aberrant XBP1 activation in tumour-associated dendritic cells led to lipid accumulation (due to aberrant triglyceride synthesis and accumulation), disrupting their normal antigen-presenting capacity, and XBP1-deficient ovarian cancer-associated DCs showed enhanced antigen-presenting capacity [
33].
This entry is adapted from the peer-reviewed paper 10.3390/genes14122118