Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Glucagon was initially regarded as a hyperglycemic substance; however, research has revealed its broader role in metabolism, encompassing effects on glucose, amino acids (AAs), and lipid metabolism. Notably, the interplay of glucagon with nutrient intake, particularly of AAs, and non-nutrient components is central to its secretion. Fasting and postprandial hyperglucagonemia have long been linked to the development and progression of type 2 diabetes (T2DM). However, studies have brought to light the positive impact of glucagon agonists on lipid metabolism and energy homeostasis.
- glucagon
- glucose
- lipid
- amino acid
- hyperglucagonemia
1. Introduction
Glucagon, a 29-amino acid peptide, was discovered in 1921 [1] and was described as a hyperglycemia substance due to contaminants in pancreatic extracts [2] in 1923. In 1948, it was established that glucagon is released from pancreatic α-cells and later, to a lesser extent, from brainstem neurons [3][4]. It is widely recognized that fasting and postprandial hyperglucagonemia play a crucial role in both the development and progression of type 2 diabetes (T2DM). However, researchers recently reshaped the role of glucagon in metabolism, confirming that the biology of glucagon is more comprehensive and extends beyond hepatic hyperglycemic actions to exert effects on glucose, amino acids (AAs), and lipid metabolism.
The hyperglycemic effect in T2DM is undisputedly present, as demonstrated by glucagon receptor antagonists in humans that, however, induced hepatic side effects [5][6][7]. The positive effects of glucagon agonists on lipid metabolism, energy homeostasis, and the reduction of liver fat have been emphasized with the development of glucagon/GLP-1 co-agonists as well as GLP-1/GIP/glucagon triple agonists, which are currently in clinical development/trials [8][9][10].
Nutrients, especially amino acids (AAs), and non-nutrient components stimulate glucagon secretion directly through sensory transporters and receptors or indirectly through their effects on cellular metabolism. Indeed, increasing protein intake, and thereby glucagon release, has shown positive effects in studies with orally treated T2DM patients [11][12].
2. Glucagon Actions and Regulation
In the decades following its discovery, glucagon was viewed primarily as the counter-regulatory hormone of insulin, governing glucose homeostasis [13][14]. Nowadays, glucagon is considered as a pleiotropic hormone whose metabolic actions include insulin secretion [15], the regulation of lipid and AA metabolism, increasing energy expenditure, the modulation of food intake and satiety, and the facilitation of weight loss in both animals and humans [16][17][18]. Furthermore, glucagon’s influence extends to the regulation of bile acid metabolism, encompassing processes such as bile acid synthesis, uptake, and efflux [19].
The secretion of glucagon by α-cells is primarily associated with occurrences of hypoglycemia, acting as a safeguard against low blood sugar levels. Additionally, it is also stimulated by elevated circulating levels of AAs and fatty acids, as well as in response to adrenergic stimulation and circulating incretins [20]. Notably, established evidence indicates that β-cell-derived secretory products, including insulin, zinc and gamma-aminobutyric acid (GABA), inhibit glucagon secretion [21]. Meanwhile, it is potently suppressed by somatostatin, GLP-1, amylin, leptin, fatty acids, and ketone bodies and stimulated by GIP and vagal stimulation. Meier et al. demonstrated that GLP-2 also stimulates glucagon secretion in healthy subjects [22] (Figure 1). And some medications such as furosemide or acetylsalicylic acid may influence prostaglandin (PG) synthesis, mainly PGE, which in turn controls glucagon release. In this regard, details of glucagon receptor function are helpful to understand its link to the metabolic effects of glucagon.
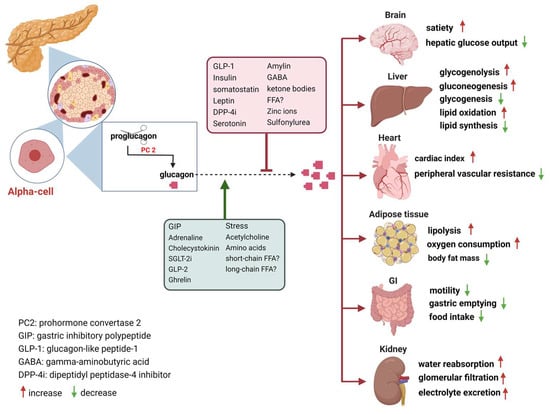
Figure 1. Glucagon receptors on multiple organs and the stimulation/inhibition of glucagon release. (This graph was generated with www.biorender.de).
How Are GCGRs Regulated?
Glucagon receptors (GCGRs) belong to the class B of seven-transmembrane (7TM) protein receptors known to activate adenylyl cyclase through Gαs-coupled proteins, which is accompanied by an increase in cellular cyclic AMP levels and activation of protein kinase A (PKA) [23][24]. Recently, GCGRs were also shown to activate the IP3 pathway via Gq and the activation of the inositol triphosphate receptor 1 (INSP3R1) in liver cells [25]. GCGRs are highly expressed in the liver as well as in multiple extrahepatic tissues (Figure 1), which play an essential role in glucose, AA, lipid, and energy metabolism [21][26][27].
Downstream signaling pathways of GCGRs involve the activation of Gs and Gq, leading to the formation of intracellular cAMP and inositol 1,4,5-trisphosphate (IP3), with the subsequent release of intracellular Ca2+. Activation of either Gs-induced protein kinase A (PKA) or Gq’s effect on Ca2+/calmodulin-dependent protein kinase can lead to the phosphorylation of the cAMP-response-element-binding protein (CREB). The metabolic effects of glucagon depend on its concentration, spatial features (mitochondrial vs. cytosolic effects), and substrate dependency. Hence, optimal plasma concentrations of substrates, specifically amino acids (AAs) and free fatty acids, play a pivotal role in both ureagenesis and gluconeogenesis processes. Prior investigations have elucidated that INSP3R1 is the isoform primarily responsible for mitochondrial calcium signaling in hepatocytes, and knocking down INSP3R1 reduced glucose production in isolated hepatocytes [28][29].
3. Glucagon and Glucose Metabolism
Glucose inhibits glucagon release upon oral or i.v. administration, while hypoglycemia increases the secretion of glucagon to elevate hepatic glucose output by stimulating glycogenolysis and gluconeogenesis, and it additionally inhibits glycogenesis and glycolysis and induces ketone production through multiple mechanisms, thereby protecting against hypoglycemia [23][30][31]. Glucagon acts in concert with cortisol, growth hormone, and adrenergic hormones, which also increase hepatic glucose output in hypoglycemia. Remarkably, the blockade of GCGRs does not impair the counter-regulation against hypoglycemia [7][32][33].
Although the level of glucagon rises rapidly in the early stage of fasting, circulating glucagon concentrations drop to postprandial levels upon prolonged fasting (see below), with persistently decreasing glycemia due to glycogen depletion [34]. Glucagon physiologically regulates the early phases of fasting in non-diabetic animals and humans, which is relevant in real life since prolonged fasting is an unusual state in industrialized societies.
The inhibition of glucagon by increases in blood glucose is important to control blood glucose and is therefore finely tuned with insulin release. However, with the development of impaired glucose tolerance and T2DM, glucagon continues to induce glucose production during hyperglycemia in fasted and preprandial conditions [35][36][37]. Both fasting and postprandial hyperglucagonemia have been proposed to trigger metabolic disturbances in obese and/or prediabetic subjects [38][39].
3.1. What Are the Explanations for This Difference (Oral vs. i.v. Glucose)?
A meticulous comparison of oral or i.v. glucose administration in healthy individuals revealed a more pronounced suppression of glucagon by i.v. compared with oral glucose [40]. Given that i.v. glucose lacks the capacity to elicit incretin responses, this cannot be explained by suppression of glucagon by GLP-1. However, as GIP was stimulated by oral but not i.v. glucose, one might postulate that GIP stimulated glucagon and thereby attenuated the effect of glucose, possibly due to the glucagonotropic action of GLP-2 [40]. In people with T2DM, an i.v. glucose dose dependently suppressed glucagon even at elevated basal plasma glucose levels, while oral glucose caused an initial stimulation of glucagon that was not explained by the levels of incretin hormones [41][42]. Complementary evidence has arisen from studies conducted on isolated α-cells and pancreatic islets. Notably, glucose unexpectedly stimulates glucagon release in isolated α-cells via mechanisms which involve KATP channels and Ca2+-mediated depolarization [43], highlighting potential indirect and paracrine mechanisms governing glucagon release inhibition in vivo. In addition, glucose inhibits glucagon release in intact mouse and human islets, apparently by paracrine mechanisms involving somatostatin release that have lost their function in islets from diabetic patients [44][45][46]. This would suggest that the deficient stimulation of somatostatin release by glucose or insulin from delta-cells in islets accounts for the hyperglucagonemia in diabetic conditions. Meanwhile, alternative somatostatin-independent mechanisms have also been proposed [46][47][48].
3.2. How Does Hypoglycemia Stimulate Glucagon Secretion?
Normally, hypoglycemia triggers a counter-regulatory response in the α-cells which does not happen in many T1DM and some T2DM patients. The comprehensive mechanisms by which glucose regulates glucagon secretion remain unclear. It has been claimed that CNS and hepatoportal sensors (i.e., hypoglycemia-activated gastrointestinal neurons in the brainstem and in several hypothalamic nuclei) contribute to the control of glucagon [49][50]. Recent studies also questioned whether the primary role of glucagon is solely to elevate glucose concentrations [51]. During the onset of fasting, there is an initial surge in glucagon levels along with a decrease in blood glucose. In prolonged fasts exceeding three days, a gradual reduction in circulating glucagon levels emerges. Surprisingly, these levels eventually normalize to what is typically seen after a meal, even in the presence of consistently low blood glucose [34].
The reason why counter-regulation fails in diabetic patients is not fully understood. Interestingly, inhibiting mitochondrial ATP production or pharmacologically activating KATP channels with diazoxide mimics the dysregulation of glucagon secretion [52]. These observations collectively suggest that the glucagon secretion defect in diabetic patients may stem from disrupted mitochondrial metabolism, though the exact mechanisms remain unclear.
It has been proposed that glucagon increases with the onset of obesity and fatty liver as a consequence of hepatic glucagon resistance [53] and with insulin resistance due to the inappropriate regulation of glucagon by fasting and a static glucagon/insulin ratio [54].
4. Glucagon and Amino Acid Metabolism
In addition to its established glucose-regulatory effects, glucagon powerfully regulates hepatic AA turnover by increasing the activities of necessary transporters and enzymes in the urea cycle through cAMP–PKA–CREB signaling [55][56]. In fact, there is evidence to suggest that the impact of glucagon signaling may vary between fasting and postprandial conditions [57]. Glucagon activates the transcription of AA transporters located on the hepatocyte membrane, thus allowing increased AA uptake and substrate availability for ureagenesis [58]. In turn, AAs enhance glucagon secretion from α-cells [59]. This generates a glucagon and AA feedback loop, referred to as the “liver–α-cell axis”, which might be as important for metabolism as the glucagon–glucose loop [60][61][62][63][64]. Genetic interventions, GCGR-antibody or pharmacological inhibition of glucagon signaling leads to α-cell hypersecretion and hyperplasia as well as a decrease in the hepatic AA transporters and gene expression involved in AA metabolism, resulting in dramatically increased plasma concentrations of some but not all AAs [60][63][65]. Notably, decreased AA levels are linked to reduced target of rapamycin (mTOR) signaling in α-cells and suppressed α-cell proliferation [66]. Hormonally, protein-rich meals prompt glucagon secretion contingent on AA plasma level kinetics [67][68]. If excess AAs, i.e., more than can be utilized for protein synthesis, are taken up with meals or liberated by proteolysis upon fasting, they are used as energy substrates [69]. Since muscle and other organs lack the capacity to manage amino groups, leading to deamination of the AAs and the carbon moiety being taken for the Krebs cycle, and alanine is primarily used to shuttle the amino groups to the liver. Alanine therefore predominates within the 15 glucogenic AAs [70] and is preferentially taken up by the liver in the presence of elevated glucagon for glucose production (known as the glucose–alanine cycle or Cahill cycle) [71][72][73]. This cycle is dysregulated in dysglycemia in humans with obesity and T2DM, as exemplified by heightened splanchnic (that is, viscera and liver) alanine uptake. One recent study demonstrated that alanine transport and aminotransferase (ALT) isoform expression (ALT and ALT2) were remarkably higher in obese, prediabetes, and overtly diabetic mouse models and in individuals with metabolic diseases.
Glucagon’s influence extends beyond ureagenesis, encompassing renal nitrogen excretion [67]. Glucagon affects fluid and solute transport in the distal tubule and collecting duct by increasing hepatic cAMP secretion, which, in turn, influences the proximal tubule reabsorption of urea. This interaction increases the fractional excretion of urea, sodium, potassium, and phosphates. After oral protein loading, there was a significant correlation between GFR and the urinary urea nitrogen excretion rate [67]. Intriguingly, branched chain amino acids (BCAA) do not induce an increase in renal hemodynamics [74]. A postprandial increase in plasma glucagon could potentially counteract AA- and insulin-stimulated mTORC1 activation, leading to the suppression of protein synthesis in the liver. After ingesting a protein-rich diet, the liver shows increased rates of translation initiation and protein synthesis compared with fasted animals. A hypothesis posits that glucagon resistance, a molecular phenomenon impacting glucagon’s physiological effects on glucose, amino acid (AA), and lipid metabolism, might contribute to the development of T2DM and metabolic diseases [68].
Most circulating AAs have been shown to potently stimulate both glucagon and insulin secretion in animals and humans, albeit with varying effects among distinct AAs [75][76]. This augmented glucagon release is believed to prevent hypoglycemia after protein intake, as AAs also stimulate insulin secretion. As early as the 1970s, Unger had found that alanine infusion induced increased glucagon secretion with minimal impact on insulin in dogs. Lysine contributed to a lesser extent to α-cell secretion while BCAAs had no effects on glucagon secretion, whereas they elicited a significant insulin response [75][77][78][79]. Nevertheless, other studies have reported that BCAAs stimulate the secretion of both insulin and glucagon, particularly with oral administration, resulting in greater and more prolonged secretion of both hormones [80][81]. Later in 1974, arginine was proven to enhance both insulin and glucagon secretion, which support separate glucose and arginine receptors on both α- and β-cells in rodents, or directly promotes plasma membrane depolarization and Ca2+ influx in the α-cell [82][83].
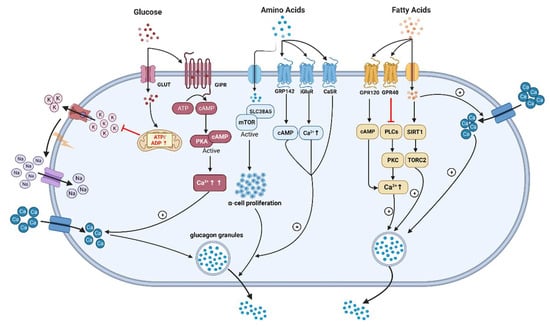
5. Glucagon and Lipid Metabolism
Glucagon is also recognized for its potent hypolipidemic effects. In humans, intravenous glucagon administration reduces the amount of plasma cholesterol, total esterified fatty acids, and apolipoproteins and the hepatic synthesis of triglycerides by stimulating β-oxidation and lipolysis in the liver [84][85]. It has been shown that glucagon can modulate the expression and activity of peroxisome proliferator-activated receptors (PPARs), affecting various aspects of lipid metabolism [86]. Glucagon’s stimulation leads to the activation of PPARα, a subtype that plays a central role in fatty acid oxidation and lipid catabolism. This interaction enhances the breakdown of fatty acids and promotes their utilization as an energy source. Meanwhile, glucagon’s influence on PPARγ affects adipocyte differentiation and insulin sensitivity. This is crucial in the context of lipid metabolism as PPARγ controls genes related to adipogenesis and lipid storage. The interplay between glucagon and PPARs highlights the complex regulatory network that orchestrates lipid utilization and storage in response to varying metabolic demands. Furthermore, glucagon also reduces hepatic lipid accumulation and decreases hepatic lipid secretion through the inhibition of lipogenesis in the liver [87]. Glucagon inhibits the activity of acetyl-CoA carboxylase, a key enzyme that initiates fatty acid synthesis by converting acetyl-CoA to malonyl-CoA. This inhibition reduces the availability of malonyl-CoA, subsequently lowering fatty acid synthesis [88].
It is established that increased GCGR signaling has been linked to improved lipid metabolism. In 1979, studies explored glucagon’s role in the direct short-term regulation of hepatic free fatty acid (FFA) metabolism, which showed that physiological concentrations of glucagon increased ketogenesis and reduced triglyceride synthesis from palmitate in hepatocytes of rats fed at FFA concentrations of 1.0 mM or lower [89]. The intricate modulation of FFA metabolism by glucagon transpires through a dual mechanism involving both intrahepatic and extrahepatic pathways
As GCGRs are expressed on β-cells and may stimulate insulin through both GLP-1R and GCGR, one may speculate that intra-islet regulation of insulin by glucagon might contribute to its effect on lipid metabolism. As discussed above, GCGR antagonists (e.g., LY2409021, Volagidemab) have been considered as glucose-lowering therapy in T2DM patients, but these resulted in lipid disorders, whereas glucagon/GLP-1 receptors co-agonism improved dyslipidemia and reduced hepatic steatosis, which have brought up discussions regarding to the relationship between glucagon signaling and lipid metabolism [5][90][91].
It remains unclear how glucagon promotes hepatic mitochondrial fat oxidation and to what extent glucagon influences lipolysis in adipose tissue, especially in humans. A previous study confirmed that INSP3R1 is essential due to the reduced glucose production seen when INSP3R1 expression was knocked down in isolated hepatocytes [29]. A recent groundbreaking discovery from Perry and co-workers [25] is quite impressive, who reported that glucagon stimulates intrahepatic lipolysis through INSP3R1/CAMKII-dependent activation with increased hepatic acetyl-CoA.
In turn, the capability of FFAs to regulate glucagon secretion remains debatable, although they are insulin secretagogues under some circumstances and increased FFAs levels might be correlated with T2DM [21][92][93][94]. Early research in 1974 had shown that the elevation of plasma FFAs suppressed glucagon levels in people, which is supported by the following clinical studies [93][95][96]. Experiments carried out on isolated rodent islets, an α-cell line, and human islets have shown that FFAs (oleate or palmitate) stimulate glucagon secretion. This occurs through signaling via fatty acid G-protein-coupled receptors, the β-oxidation of fatty acids, and the activation of L-type Ca2+ channels.
Additionally, it involves relieving the inhibitory paracrine action of somatostatin secreted from δ-cells [97][98][99]. Wang et al. reported that long-chain FFA (linoleic acid) acutely stimulated glucagon secretion by activation of G-protein-coupled receptor 40 (GPR40) and phospholipase C to increase Ca2+ release and the associated Ca2+ influx through Ca2+ channels in primary cultured rat pancreatic islets [100] (Figure 2).
Figure 2. The underlying mechanisms of glucagon stimulation to glucose, amino acids, and fatty acids (↑increase). (This graph is generated with www.biorender.de).
This entry is adapted from the peer-reviewed paper 10.3390/nu15183913
References
- Ricketts, H.T. Glucagon: Molecular Physiology, Clinical and Therapeutic Implications. JAMA 1973, 224, 403.
- Murlin, J.R.; Clough, H.D.; Gibbs, C.B.F.; Stokes, A.M. Aqueous Extracts of Pancreas. J. Biol. Chem. 1923, 56, 253–296.
- Sutherland, E.W.; de Duve, C. Origin and Distribution of the Hyperglycemic-Glycogenolytic Factor of the Pancreas. J. Biol. Chem. 1948, 175, 663–674.
- Sasaki, H.; Ebitani, I.; Tominaga, M.; Yamatani, K.; Yawata, Y.; Hara, M. Glucagon-like substance in the canine brain. Endocrinol. Jpn. 1980, 27 (Suppl. 1), 135–140.
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T.A. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528.
- Pettus, J.H.; D’Alessio, D.; Frias, J.P.; Vajda, E.G.; Pipkin, J.D.; Rosenstock, J.; Williamson, G.; Zangmeister, M.A.; Zhi, L.; Marschke, K.B. Efficacy and Safety of the Glucagon Receptor Antagonist RVT-1502 in Type 2 Diabetes Uncontrolled on Metformin Monotherapy: A 12-Week Dose-Ranging Study. Diabetes Care 2020, 43, 161–168.
- Kazierad, D.J.; Chidsey, K.; Somayaji, V.R.; Bergman, A.J.; Calle, R.A. Efficacy and safety of the glucagon receptor antagonist PF-06291874: A 12-week, randomized, dose-response study in patients with type 2 diabetes mellitus on background metformin therapy. Diabetes Obes. Metab. 2018, 20, 2608–2616.
- Bossart, M.; Wagner, M.; Elvert, R.; Evers, A.; Hubschle, T.; Kloeckener, T.; Lorenz, K.; Moessinger, C.; Eriksson, O.; Velikyan, I.; et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022, 34, 59–74 e10.
- Finan, B.; Capozzi, M.E.; Campbell, J.E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 2020, 69, 532–541.
- Rosenstock, J.; Wysham, C.; Frias, J.P.; Kaneko, S.; Lee, C.J.; Fernandez Lando, L.; Mao, H.; Cui, X.; Karanikas, C.A.; Thieu, V.T. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): A double-blind, randomised, phase 3 trial. Lancet 2021, 398, 143–155.
- Sucher, S.; Markova, M.; Hornemann, S.; Pivovarova, O.; Rudovich, N.; Thomann, R.; Schneeweiss, R.; Rohn, S.; Pfeiffer, A.F.H. Comparison of the effects of diets high in animal or plant protein on metabolic and cardiovascular markers in type 2 diabetes: A randomized clinical trial. Diabetes Obes. Metab. 2017, 19, 944–952.
- Markova, M.; Pivovarova, O.; Hornemann, S.; Sucher, S.; Frahnow, T.; Wegner, K.; Machann, J.; Petzke, K.J.; Hierholzer, J.; Lichtinghagen, R.; et al. Isocaloric Diets High in Animal or Plant Protein Reduce Liver Fat and Inflammation in Individuals With Type 2 Diabetes. Gastroenterology 2017, 152, 571–585 e578.
- Unger, R.H.; Orci, L. Paracrinology of islets and the paracrinopathy of diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 16009–16012.
- Sherwin, R.; Felig, P. Glucagon physiology in health and disease. Int. Rev. Physiol. 1977, 16, 151–171.
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when beta-cells are active. JCI Insight 2019, 5, 129954.
- Schulman, J.L.; Carleton, J.L.; Whitney, G.; Whitehorn, J.C. Effect of glucagon on food intake and body weight in man. J. Appl. Physiol. 1957, 11, 419–421.
- Billington, D.C. Angiogenesis and its inhibition: Potential new therapies in oncology and non-neoplastic diseases. Drug Des. Discov. 1991, 8, 3–35.
- Geary, N.; Kissileff, H.R.; Pi-Sunyer, F.X.; Hinton, V. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am. J. Physiol. Integr. Comp. Physiol. 1992, 262, R975–R980.
- Bouscarel, B.; Kroll, S.D.; Fromm, H. Signal transduction and hepatocellular bile acid transport: Cross talk between bile acids and second messengers. Gastroenterology 1999, 117, 433–452.
- Holst, J.J.; Knop, F.K.; Vilsboll, T.; Krarup, T.; Madsbad, S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 2011, 34 (Suppl. 2), S251–S257.
- Gromada, J.; Franklin, I.; Wollheim, C.B. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007, 28, 84–116.
- Meier, J.J.; Nauck, M.A.; Pott, A.; Heinze, K.; Goetze, O.; Bulut, K.; Schmidt, W.E.; Gallwitz, B.; Holst, J.J. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology 2006, 130, 44–54.
- Habegger, K.M.; Heppner, K.M.; Geary, N.; Bartness, T.J.; DiMarchi, R.; Tschop, M.H. The metabolic actions of glucagon revisited. Nat. Rev. Endocrinol. 2010, 6, 689–697.
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13 (Suppl. 1), 118–125.
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283.
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837.
- Bansal, P.; Wang, Q. Insulin as a physiological modulator of glucagon secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E751–E761.
- Feriod, C.N.; Oliveira, A.G.; Guerra, M.T.; Nguyen, L.; Richards, K.M.; Jurczak, M.J.; Ruan, H.B.; Camporez, J.P.; Yang, X.; Shulman, G.I.; et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol. Commun. 2017, 1, 23–35.
- Wang, Y.; Li, G.; Goode, J.; Paz, J.C.; Ouyang, K.; Screaton, R.; Fischer, W.H.; Chen, J.; Tabas, I.; Montminy, M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 2012, 485, 128–132.
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678.
- Sokal, J.E. Effect of glucagon on gluconeogenesis by the isolated perfused rat liver. Endocrinology 1966, 78, 538–548.
- Gerich, J.E.; Lorenzi, M.; Bier, D.M.; Tsalikian, E.; Schneider, V.; Karam, J.H.; Forsham, P.H. Effects of physiologic levels of glucagon and growth hormone on human carbohydrate and lipid metabolism. Studies involving administration of exogenous hormone during suppression of endogenous hormone secretion with somatostatin. J. Clin. Investig. 1976, 57, 875–884.
- Lecavalier, L.; Bolli, G.; Gerich, J. Glucagon-cortisol interactions on glucose turnover and lactate gluconeogenesis in normal humans. Am. J. Physiol. 1990, 258, E569–E575.
- Marliss, E.B.; Aoki, T.T.; Unger, R.H.; Soeldner, J.S.; Cahill, G.F., Jr. Glucagon levels and metabolic effects in fasting man. J. Clin. Investig. 1970, 49, 2256–2270.
- Rohrer, S.; Menge, B.A.; Gruber, L.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; Holst, J.J.; Meier, J.J. Impaired crosstalk between pulsatile insulin and glucagon secretion in prediabetic individuals. J. Clin. Endocrinol. Metab. 2012, 97, E791–E795.
- Menge, B.A.; Gruber, L.; Jorgensen, S.M.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; Holst, J.J.; Meier, J.J. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 2011, 60, 2160–2168.
- Meier, J.J.; Kjems, L.L.; Veldhuis, J.D.; Lefebvre, P.; Butler, P.C. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: Further evidence for the intraislet insulin hypothesis. Diabetes 2006, 55, 1051–1056.
- Demant, M.; Bagger, J.I.; Suppli, M.P.; Lund, A.; Gyldenlove, M.; Hansen, K.B.; Hare, K.J.; Christensen, M.; Sonne, D.P.; Holst, J.J.; et al. Determinants of Fasting Hyperglucagonemia in Patients with Type 2 Diabetes and Nondiabetic Control Subjects. Metab. Syndr. Relat. Disord. 2018, 16, 530–536.
- Lee, Y.H.; Wang, M.-Y.; Yu, X.-X.; Unger, R.H. Glucagon is the key factor in the development of diabetes. Diabetologia 2016, 59, 1372–1375.
- Meier, J.J.; Deacon, C.F.; Schmidt, W.E.; Holst, J.J.; Nauck, M.A. Suppression of glucagon secretion is lower after oral glucose administration than during intravenous glucose administration in human subjects. Diabetologia 2007, 50, 806–813.
- Bagger, J.I.; Knop, F.K.; Lund, A.; Holst, J.J.; Vilsboll, T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia 2014, 57, 1720–1725.
- Bagger, J.I.; Knop, F.K.; Lund, A.; Vestergaard, H.; Holst, J.J.; Vilsboll, T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 737–745.
- Olsen, H.L.; Theander, S.; Bokvist, K.; Buschard, K.; Wollheim, C.B.; Gromada, J. Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells. Endocrinology 2005, 146, 4861–4870.
- Omar-Hmeadi, M.; Lund, P.E.; Gandasi, N.R.; Tengholm, A.; Barg, S. Paracrine control of alpha-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat. Commun. 2020, 11, 1896.
- Kellard, J.A.; Rorsman, N.J.G.; Hill, T.G.; Armour, S.L.; van de Bunt, M.; Rorsman, P.; Knudsen, J.G.; Briant, L.J.B. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol. Metab. 2020, 40, 101021.
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139.
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224.
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291.
- Steinbusch, L.; Labouebe, G.; Thorens, B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol. Metab. 2015, 26, 455–466.
- Stanley, S.; Moheet, A.; Seaquist, E.R. Central Mechanisms of Glucose Sensing and Counterregulation in Defense of Hypoglycemia. Endocr. Rev. 2019, 40, 768–788.
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536.
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882.
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The Liver-alpha-Cell Axis and Type 2 Diabetes. Endocr. Rev. 2019, 40, 1353–1366.
- Stern, J.H.; Smith, G.I.; Chen, S.; Unger, R.H.; Klein, S.; Scherer, P.E. Obesity dysregulates fasting-induced changes in glucagon secretion. J. Endocrinol. 2019, 243, 149–160.
- Snodgrass, P.J.; Lin, R.C.; Muller, W.A.; Aoki, T.T. Induction of urea cycle enzymes of rat liver by glucagon. J. Biol. Chem. 1978, 253, 2748–2753.
- Hamberg, O.; Vilstrup, H. Regulation of urea synthesis by glucose and glucagon in normal man. Clin. Nutr. 1994, 13, 183–191.
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; Fu, H.; Watson, D.E.; Lewin, A.J.; Landschulz, W.H.; et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care 2016, 39, 1241–1249.
- Kilberg, M.S.; Barber, E.F.; Handlogten, M.E. Characteristics and hormonal regulation of amino acid transport system A in isolated rat hepatocytes. Curr. Top. Cell Regul. 1985, 25, 133–163.
- Ohneda, A.; Parada, E.; Eisentraut, A.M.; Unger, R.H. Characterization of response of circulating glucagon to intraduodenal and intravenous administration of amino acids. J. Clin. Investig. 1968, 47, 2305–2322.
- Dean, E.D. A Primary Role for α-Cells as Amino Acid Sensors. Diabetes 2020, 69, 542–549.
- Dean, E.D.; Unger, R.H.; Holland, W.L. Glucagon antagonism in islet cell proliferation. Proc. Natl. Acad. Sci. USA 2017, 114, 3006–3008.
- Flakoll, P.J.; Borel, M.J.; Wentzel, L.S.; Williams, P.E.; Lacy, D.B.; Abumrad, N.N. The role of glucagon in the control of protein and amino acid metabolism in vivo. Metabolism 1994, 43, 1509–1516.
- Boden, G.; Rezvani, I.; Owen, O.E. Effects of glucagon on plasma amino acids. J. Clin. Investig. 1984, 73, 785–793.
- Holst, J.J.; Wewer Albrechtsen, N.J.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver-alpha-Cell Axis. Diabetes 2017, 66, 235–240.
- Miller, R.A.; Birnbaum, M.J. Glucagon: Acute actions on hepatic metabolism. Diabetologia 2016, 59, 1376–1381.
- Solloway, M.J.; Madjidi, A.; Gu, C.; Eastham-Anderson, J.; Clarke, H.J.; Kljavin, N.; Zavala-Solorio, J.; Kates, L.; Friedman, B.; Brauer, M.; et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of alpha-Cell Mass. Cell Rep. 2015, 12, 495–510.
- Bankir, L.; Bouby, N.; Speth, R.C.; Velho, G.; Crambert, G. Glucagon revisited: Coordinated actions on the liver and kidney. Diabetes Res. Clin. Pr. 2018, 146, 119–129.
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sorensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314.
- Koopman, R.; Walrand, S.; Beelen, M.; Gijsen, A.P.; Kies, A.K.; Boirie, Y.; Saris, W.H.; van Loon, L.J. Dietary protein digestion and absorption rates and the subsequent postprandial muscle protein synthetic response do not differ between young and elderly men. J. Nutr. 2009, 139, 1707–1713.
- Wahren, J.; Ekberg, K. Splanchnic regulation of glucose production. Annu. Rev. Nutr. 2007, 27, 329–345.
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207.
- Okun, J.G.; Rusu, P.M.; Chan, A.Y.; Wu, Y.; Yap, Y.W.; Sharkie, T.; Schumacher, J.; Schmidt, K.V.; Roberts-Thomson, K.M.; Russell, R.D.; et al. Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat. Metab. 2021, 3, 394–409.
- Snell, K.; Duff, D.A. The hepato-muscular metabolic axis and gluconeogenesis. Prog. Clin. Biol. Res. 1982, 102 Pt C, 279–291.
- Claris-Appiani, A.; Assael, B.M.; Tirelli, A.S.; Marra, G.; Cavanna, G. Lack of glomerular hemodynamic stimulation after infusion of branched-chain amino acids. Kidney Int. 1988, 33, 91–94.
- Rocha, D.M.; Faloona, G.R.; Unger, R.H. Glucagon-stimulating activity of 20 amino acids in dogs. J. Clin. Investig. 1972, 51, 2346–2351.
- Galsgaard, K.D.; Jepsen, S.L.; Kjeldsen, S.A.S.; Pedersen, J.; Wewer Albrechtsen, N.J.; Holst, J.J. Alanine, arginine, cysteine, and proline, but not glutamine, are substrates for, and acute mediators of, the liver-alpha-cell axis in female mice. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E920–E929.
- Kaneto, A.; Kosaka, K. Effects of leucine and isoleucine infused intrapancreatically on glucagon and insulin secretion. Endocrinology 1972, 91, 691–695.
- Muller, W.A.; Faloona, G.R.; Unger, R.H. The effect of alanine on glucagon secretion. J. Clin. Investig. 1971, 50, 2215–2218.
- Kuhara, T.; Ikeda, S.; Ohneda, A.; Sasaki, Y. Effects of intravenous infusion of 17 amino acids on the secretion of GH, glucagon, and insulin in sheep. Am. J. Physiol. 1991, 260, E21–E26.
- Gar, C.; Rottenkolber, M.; Prehn, C.; Adamski, J.; Seissler, J.; Lechner, A. Serum and plasma amino acids as markers of prediabetes, insulin resistance, and incident diabetes. Crit. Rev. Clin. Lab. Sci. 2018, 55, 21–32.
- Adeva-Andany, M.M.; Lopez-Maside, L.; Donapetry-Garcia, C.; Fernandez-Fernandez, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 2017, 49, 1005–1028.
- Gerich, J.E.; Charles, M.A.; Grodsky, G.M. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J. Clin. Investig. 1974, 54, 833–841.
- Assan, R.; Attali, J.R.; Ballerio, G.; Boillot, J.; Girard, J.R. Glucagon secretion induced by natural and artificial amino acids in the perfused rat pancreas. Diabetes 1977, 26, 300–307.
- Pegorier, J.P.; Garcia-Garcia, M.V.; Prip-Buus, C.; Duee, P.H.; Kohl, C.; Girard, J. Induction of ketogenesis and fatty acid oxidation by glucagon and cyclic AMP in cultured hepatocytes from rabbit fetuses. Evidence for a decreased sensitivity of carnitine palmitoyltransferase I to malonyl-CoA inhibition after glucagon or cyclic AMP treatment. Biochem. J. 1989, 264, 93–100.
- Guettet, C.; Mathe, D.; Riottot, M.; Lutton, C. Effects of chronic glucagon administration on cholesterol and bile acid metabolism. Biochim. Biophys. Acta 1988, 963, 215–223.
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627.
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413.
- Peng, I.C.; Chen, Z.; Sun, W.; Li, Y.S.; Marin, T.L.; Hsu, P.H.; Su, M.I.; Cui, X.; Pan, S.; Lytle, C.Y.; et al. Glucagon regulates ACC activity in adipocytes through the CAMKKbeta/AMPK pathway. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1560–E1568.
- Witters, L.A.; Trasko, C.S. Regulation of hepatic free fatty acid metabolism by glucagon and insulin. Am. J. Physiol. 1979, 237, E23–E29.
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757.
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266.
- Boden, G.; Carnell, L.H. Nutritional effects of fat on carbohydrate metabolism. Best. Pr. Res. Clin. Endocrinol. Metab. 2003, 17, 399–410.
- Gerich, J.E.; Langlois, M.; Schneider, V.; Karam, J.H.; Noacco, C. Effects of alternations of plasma free fatty acid levels on pancreatic glucagon secretion in man. J. Clin. Investig. 1974, 53, 1284–1289.
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Investig. 2002, 32 (Suppl. 3), 14–23.
- Madison, L.L.; Seyffert, W.A., Jr.; Unger, R.H.; Barker, B. Effect on plasma free fatty acids on plasma glucagon and serum insulin concentrations. Metabolism 1968, 17, 301–304.
- Edwards, J.C.; Taylor, K.W. Fatty acids and the release of glucagon from isolated guinea-pig islets of Langerhans incubated in vitro. Biochim. Biophys. Acta 1970, 215, 310–315.
- Collins, S.C.; Salehi, A.; Eliasson, L.; Olofsson, C.S.; Rorsman, P. Long-term exposure of mouse pancreatic islets to oleate or palmitate results in reduced glucose-induced somatostatin and oversecretion of glucagon. Diabetologia 2008, 51, 1689–1693.
- Olofsson, C.S.; Salehi, A.; Gopel, S.O.; Holm, C.; Rorsman, P. Palmitate stimulation of glucagon secretion in mouse pancreatic alpha-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes 2004, 53, 2836–2843.
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Gopel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 4657.
- Wang, L.; Zhao, Y.; Gui, B.; Fu, R.; Ma, F.; Yu, J.; Qu, P.; Dong, L.; Chen, C. Acute stimulation of glucagon secretion by linoleic acid results from GPR40 activation and i increase in pancreatic islet -cells. J. Endocrinol. 2011, 210, 173–179.
This entry is offline, you can click here to edit this entry!