Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
|
Oncology
P-glycoprotein (P-gp) is a crucial membrane transporter situated on the cell’s apical surface, being responsible for eliminating xenobiotics and endobiotics. P-gp modulators are compounds that can directly or indirectly affect this protein, leading to changes in its expression and function. These modulators can act as inhibitors, inducers, or activators, potentially causing drug–drug interactions (DDIs).
- P-glycoprotein
1. Introduction
In therapeutics, the simultaneous administration of multiple drugs is very frequent, leading to several drug–drug interactions (DDIs), which may result in a higher or lower bioavailability of a given drug, according to the occurrence of inhibition or induction/activation of drug transporters/metabolic enzymes, respectively [1,2]. By definition, DDIs cause changes in the therapeutic/toxic effects of one compound (substrate) due to the co-administration of another compound (modulator). Moreover, DDIs are triggered by two different types of interactions: at the pharmacodynamic or pharmacokinetic levels. An example of pharmacodynamic interactions is the excessive bleeding observed after the concomitant use of warfarin (a vitamin K antagonist) and low-dose aspirin. By decreasing the production of coagulation factors, warfarin affects bleeding, an effect that is further increased by aspirin through the inhibition of thrombocyte aggregation. On the other hand, pharmacokinetic interactions can also occur due to alterations in the absorption, distribution, metabolism, and elimination (ADME) of a given drug, which may lead to higher or lower drug levels and, consequently, to potential toxicity or loss of therapeutic efficacy, respectively. These alterations may be due to interactions of xenobiotics with drug-metabolizing enzymes, such as those belonging to the cytochrome P450 system (CYP450), involved in phase I reactions, or uridine diphosphate glucuronosyltransferases, involved in phase II reactions [3]. In addition, xenobiotics can also interfere with the transporters present at the cell membranes of different tissues/organs [3,4].
Transporters are large proteins incorporated into the cell membranes, which are expressed in several cells of the major organs with functions of absorption and elimination, such as the liver, intestines, kidneys, brain, testes, and placenta—sites of high importance regarding the evaluation of the pharmacokinetics of xenobiotics [5]. Transporters control the absorption and elimination of xenobiotics and also regulate the movement of small endogenous molecules, such as key metabolites, signaling molecules, vitamins, antioxidants (e.g., uric acid), and some hormones [6]. Recently, the terms phases 0 and III, associated with the uptake and efflux transport of drugs, respectively, were added to the phases I and II that were already associated with the metabolism (functionalization or conjugation, respectively), thus characterizing the complete movement of a molecule in a given cell [7,8].
In the kidneys, uptake transporters allow the passage of the compounds from blood or ultrafiltrate into the proximal tubular epithelial cells (PTECs), while efflux transporters excrete them back into the blood or into the urine [9]. Furthermore, in the intestines, uptake transporters move the compounds from blood or the intestinal lumen into the enterocytes, while efflux transporters transport them mainly into the intestinal lumen [10,11]. In both organs, there are two superfamilies that represent the most expressed transporters: the solute carrier (SLC) transporters superfamily, which mostly ensures the cellular uptake (phase 0) of compounds from the blood, at the basolateral membrane; and the adenosine triphosphate (ATP)-binding cassette (ABC) transporters superfamily, which enables the efflux (phase III) of their substrates to the tubular/intestinal lumen, at the apical membrane [12,13]. In this way, it is important to note that the tubular secretion of a xenobiotic may involve at least one transporter of each type [13]. Figure 1 shows the major transporters involved in the uptake and/or efflux of substrates at the kidney and intestinal levels. As mentioned above, the xenobiotics’ secretion is based on a coordinated process of transporter-mediated movement across the basolateral and apical cellular membranes. Therefore, the modulation of such transporters by other compounds can potentially result in alterations in the substrate plasma concentrations [14,15].
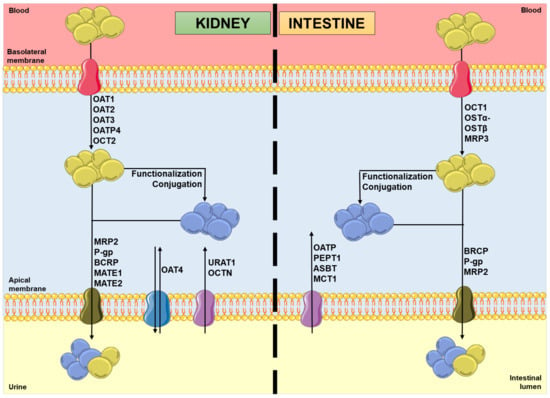
Figure 1. Schematic representation of molecules’ movement and metabolism in proximal tubular epithelial cells and enterocytes, including the transporters’ location and the direction of the substrates’ movement, involved in phases 0 and III of pharmacokinetics. Legend: ASBT: ileal apical sodium/bile acid co-transporter; BCRP: breast cancer resistance protein; blue spheres: xenobiotics; MATE: multidrug and toxin extrusion protein; MCT: monocarboxylic acid transporter; MRP: multidrug resistance protein; OAT: organic anion transporter; OATP: organic anion-transporting polypeptide; OCT: organic cation transporter; OCTN: organic cation/ergothioneine transporter; OSTα-OSTβ: heteromeric organic solute transporter; PEPT: peptide transporter; P-gp: P-glycoprotein; URAT: urate transporter; yellow spheres: xenobiotic metabolites.
Despite the undesirable and sometimes life-threatening consequences that DDIs can lead to, on the other hand, it is possible to take advantage of them as a novel therapeutic strategy and, thus, study these transporters as targets to treat diseases [16]. In this sense, the scientific community has made great progress in the predictability and modeling of DDIs [15]. The goal is to evaluate the DDI potential in order to decrease the toxicity risks and/or enhance the therapeutic effectiveness, employing combined approaches using in silico, in vitro, and/or in vivo models [17]. Focusing on the computational models, modeling of transporters may provide important insights into the proteins’ structures, membrane topology, and different conformational states, as well as increasing the knowledge of the substrate binding, translocation, and kinetics processes, and raising awareness of the mechanisms underlying modulators’ modes of action [16].
2. Overview of P-Glycoprotein
2.1. Description
P-glycoprotein (P-gp), which comes from “permeability-glycoprotein”, also called multidrug resistance protein 1 (MDR1), is expressed by the MDR1/ABCB1 gene in humans and belongs to the ABC transporters superfamily, as already mentioned [18,19]. In 1976, Juliano and Ling discovered this protein in hamster ovary cells [20]. Since then, P-gp has been a well-studied protein due to its particularly important role in protecting various sensitive tissues against different xenobiotics, by actively pumping these molecules to outside of the cells and, consequently, decreasing their intracellular concentration and toxicity. Thus, it plays an important role in the process of protection and detoxification, as well as being markedly involved in the phenomenon of multidrug resistance (MDR) in tumor chemotherapy [18,21]. In humans, there are two P-gp isoforms: MDR1 and MDR3. However, only the coding product of the MDR1/ABCB1 gene actively and significantly participates in the disposition of xenobiotics in various organs. The MDR3/ABCB4 gene encodes for a phospholipid lipase protein that is mainly expressed on the canalicular domain of hepatocytes, being responsible for the secretion of phospholipids from hepatocytes into bile [22]. In this way, the dysfunction and/or deficiency of this transporter can result in altered phosphatidylcholine–bile salt ratios and, consequently, intrahepatic cholestasis of pregnancy, low-phospholipid-associated cholelithiasis, drug-induced liver injury, or even progressive familial intrahepatic cholestasis type 3 [23].
2.2. P-gp’s Structure
This transporter is synthesized in the endoplasmic reticulum as a glycosylated intermediate with a molecular weight of 150–170 kDa, with the carbohydrate moiety being further modified in the Golgi apparatus prior to its export to the cell surface [21,24,25]. Like other ABC transporters, P-gp is considered to be a full transporter with two transmembrane domains (TMDs), consisting of six transmembrane helices (TMHs) each, where the substrates connect, and two nucleotide-binding domains (NBDs), where ATP links in order to proceed to its own hydrolysis [21,26,27,28].
Currently, a more detailed constitution of P-gp is accepted among the scientific community, where there are considered to be two homologous functional units (N- and C-terminal halves) with a pseudo-twofold symmetry, each composed of one TMD (comprising six TMHs) and one NBD (containing a catalytic site for ATP binding and hydrolysis). These N- and C-terminal halves are connected by a small peptide sequence (the “linker”), while the TMHs are directly linked to the respective NBD by the intracellular loops, through the functional TMHs 6 (NBD1) and 12 (NBD2), and non-covalently by short intracellular coupling helices (ICHs). These ICHs are located between the structural TMHs 2/3 (ICH1-NBD1), 4/5 (ICH2-NBD2), 8/9 (ICH3-NBD2), and 10/11 (ICH4-NBD1) and have important functions in the maturation and folding of the P-gp transporter, as well as being involved in the signal transmission pathways between the TMDs and NBDs. Furthermore, instead of the two distinct DBSs reported by Shapiro and Ling, (1997), there is now considered to be a large cavity formed by the TMHs of both the N- and C-terminal P-gp halves, called the drug-binding pocket (DBP), which can recognize and accommodate several structurally distinct substrates [29].
2.3. P-gp Efflux Mechanisms
There are two efflux mechanisms proposed for the P-gp-mediated transport: the flippase model, and the hydrophobic vacuum cleaner model [30,31], indicating that the extrusion of the molecules can occur directly from the cytoplasmic compartment or that the protein could act as a membrane pore [31,32]. Currently, the most accepted theory is the “hydrophobic vacuum cleaner” model, which correlates the diffusion of the transported molecules within the inner leaflet of the cellular membrane with interactions at the drug-binding site (DBS) [31,33]. In the late 1990s, Shapiro and Ling proposed the existence of two distinct DBSs, positively cooperating with one another, which were named the H-site and R-site due to the distinct drug specificities registered for Hoechst 33342 and rhodamine-123 (RHO123), respectively [34]. Moreover, it was also shown that molecules could be removed from the inner leaflet to the outer leaflet of the lipid bilayer through both sites, supporting the vacuum cleaner model and postulating the existence of two distinct translocation pathways [31,34,35]. However, several doubts remain today about the specific location of the DBS [31].
P-gp undergoes dynamic conformational changes, through which DBSs alternately access the two sides of the membrane, connecting a charge on one side and releasing it on the other [36,37]. The membrane transporter’s work depends on the energy resulting from the ATP hydrolysis. It has been suggested that the binding of ATP to the NBD and the dimerization of the NBD are driving forces for this function. The hydrolysis energy causes complete conformational changes in the TMD and catalyzes the efflux of the substrate through the TMD and the lipid bilayer. Thereafter, a redefinition of the membrane transporter to its original conformation occurs, allowing new catalytic cycles to start [33,36]. However, the mechanism of communication between the DBS and NBD, along with the extent of the conformational changes leading to the efflux mechanism, remains not fully understood. Although the crystallographic structure of P-gp can be used as a framework to allow a better understanding of its mechanism of action, it has been proven that this study is challenging, significantly decreasing the number of available structures in the Protein Data Bank (PDB) [33].
This transporter exports a broad range of structurally unrelated compounds, being a non-specific transporter [26,29]. P-gp shares some substrates with other ABC transporters, including multidrug resistance protein 1 (MRP1) and multidrug resistance protein 2 (MRP2), more specifically cytostatic agents [26]. P-gp substrates vary greatly in size, structure, and function, ranging from small molecules, such as organic cations, carbohydrates, amino acids, and some antibiotics, to macromolecules, such as polysaccharides and proteins, which is consistent with the knowledge that P-gp is a protective protein against several compounds [24]. Furthermore, several drugs used in therapy are considered to be P-gp substrates, including anticancer agents, cardiac glycosides (e.g., digoxin), β-adrenoceptor antagonists, Ca2+ channel blockers, HIV protease inhibitors, steroids, immunosuppressants, antiemetic drugs, antibiotics, antimicrobials, antiretrovirals, and histamine H1 receptor antagonists [7,13,24,38,39].
2.4. P-gp Expression
P-gp has low expression in most human tissues, but it is found at much higher expression levels in epithelial cells of the intestine (apical membrane of the enterocytes in the lower gastrointestinal tract—jejunum, duodenum, ileum, and colon), limiting the absorption of drug substrates from the gastrointestinal tract; bile ducts of the liver and proximal kidney tubules, resulting in improved excretion of drug substrates into the bile and urine, respectively; the blood–brain barrier (BBB), limiting the entry of a variety of xenobiotics and/or their metabolites into the central nervous system (CNS); hematopoietic cells (where its function remains unknown); and other sites, such as the pancreatic ducts, adrenal glands, placenta, endometrium, and testicles. In general, the location of this protein reinforces the accomplishment of its protective/barrier function. Its physiological role in secreting endogenous and exogenous compounds (through phase III-mediated transport) influences the (pharmaco)toxicokinetics of several compounds [21,22,25,40]. This is what happens in the so-called “normal” cells under physiological conditions. However, in tumor cells, there is frequently an overexpression of P-gp, which causes anticancer agents to be exported from the tumor cells. This phenomenon keeps the concentrations of these drugs below the therapeutic threshold, which can lead to a subtherapeutic effect on these cells, thus creating tumor resistance to these types of drug agents [36].
Additionally, polymorphisms affecting the MDR1/ABCB1 gene should also be considered. Indeed, MDR1/ABCB1 polymorphisms are associated with lower renal digoxin clearance, as well as modified efficacy and nephrotoxicity of chemotherapeutics, antivirals, and immunosuppressants, like cyclosporine (due to impaired apical efflux) [13]. In addition to these effects, MDR1/ABCB1 polymorphisms can also contribute to the alteration of the drugs’ disposition and, consequently, to interindividual variability in response to several drugs, like morphine and other opioids (e.g., methadone) [41]. Some authors have studied the polymorphisms of the P-gp-coding gene. Hoffmeyer et al. observed a significant correlation between a P-gp polymorphism (C3435T in exon 26) and the levels of P-gp’s expression and function. Individuals who were homozygous for this polymorphism (24% of the sample population (n = 5188)) had significantly lower duodenal P-gp expression and the highest digoxin plasma levels, suggesting that this polymorphism may affect the absorption and tissue concentrations of numerous other P-gp substrates [42]. In addition, Saiz-Rodríguez et al. performed clinical trials with 473 healthy volunteers to analyze the C3435T polymorphism, concluding that it can affect the elimination of some drugs in different ways. They observed an enhanced elimination of risperidone, trazodone, and dehydroaripiprazole, while in the cases of olanzapine and citalopram a reduction in their elimination occurred. On the other hand, there were no changes detected in the elimination of quetiapine, aripiprazole, sertraline, or agomelatine [43]. Kim et al. carried out a polymorphism analysis of 37 healthy European-American and 23 healthy African-American subjects, identifying 10 single-nucleotide polymorphisms (SNPs). Three SNPs (C1236T in exon 12; C3435T in exon 26; G2677T, Ala893Ser in exon 21) were found to be linked (MDR1*2 allele, which is suggested to cause enhanced P-gp activity) and occurred in 62% of European Americans and 13% of African Americans. In vitro experiments indicated an enhanced efflux of digoxin (50 µmol/L) from cells expressing the MDR1-Ser893 variant (HEK293T cells, human embryonic kidney 293 (HEK293) containing simian vacuolating virus 40 (SV40) large T antigen). In humans, MDR1*1 and MDR1*2 variants were associated with differences in the plasma levels of fexofenadine (a known substrate of P-gp); these results were consistent with the in vitro data, suggesting that, in vivo, P-gp activity in subjects with the MDR1*2 allele is also enhanced [44].
Therefore, a deeper understanding of P-gp’s structure and efflux transport mechanism is fundamental for investigating the phenomena of modulating the activity of this transporter and the consequent modification of the (pharmaco)toxicokinetics of several xenobiotics [29].
This entry is adapted from the peer-reviewed paper 10.3390/molecules28227532
This entry is offline, you can click here to edit this entry!