Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
C-Reactive Protein (CRP) is supposed to be an accredited benchmark for physicians, to reveal or rule out inflammation, and multifarious scientific endeavors have been made so as to detect the direct pleiotropic functions of this protein. The use of CRP as the most important and critical immunochemical marker of several medical conditions, including infections such as sepsis, physiological organ diseases, various autoimmune disorders, malignancies and other health conditions, has become widely popular
- C-Reactive Protein
- CRP
- biochemical properties
- CRP forms
- CRP structure
- CRP genetics
1. Introduction
Over the last few years, multifarious conventional and point-of-care molecular diagnostic assays have shaped the accuracy of medical diagnosis to a great extent. Nowadays, numerous hematological, biochemical and serological diagnostic tests are widely performed on various clinical specimens in order to estimate the functional capacity of several critical organs and systems, identify the presence of foreign agents, and monitor the course of various (auto)immune processes and the overall immune status of a case [1,2,3,4,5,6,7,8,9,10].
C-Reactive Protein (CRP) is supposed to be an accredited benchmark for physicians, to reveal or rule out inflammation, and multifarious scientific endeavors have been made so as to detect the direct pleiotropic functions of this protein. The use of CRP as the most important and critical immunochemical marker of several medical conditions, including infections such as sepsis, physiological organ diseases, various autoimmune disorders, malignancies and other health conditions, has become widely popular [11,12,13,14,15,16,17,18,19,20]. Hitherto, a plethora of molecular diagnostic assays have been developed for the detection of CRP [21,22].
2. Current Evidence on Potential Biochemical Properties and Genetics of C-Reactive Protein
2.1. Forms of C-Reactive Protein
CRP was discovered by Tillett and Francis of Rockefeller University in 1930; they reported a non-protein somatic fraction called “fraction c” that precipitated in high titers after isolation from the serum of patients infected with pneumococcus, which was biochemically distinct from previously known capsular polysaccharide and nucleoprotein fractions detectable by a specific antibody response [23]. About a decade later, Avery and McCarty reported a substance elevated in the serum of cases with a pathogenic spectrum of inflammatory stimuli [24]. The name “C-Reactive-Protein” arose by virtue of further research by Volanakis and Kaplan, who identified the precise ligand for CRP in the pneumococcal “c” polysaccharide as phosphocholine, which is derived from the teichoic acid of the pneumococcal cell wall [25].
Phylogenetically, CRP is highly conserved with homologues in various vertebrates and invertebrates. Various physicochemical and immunological research studies on the tertiary and quaternary structure of CRP have concluded that the microenvironment can modify its architecture. Heretofore, with the exception of genetic variations, it has been revealed that CRP can exist in at least three main distinct forms, including a monomeric CRP form, often called “modified CRP”, that consists of a unique subunit; a “native” pentameric form; and a multimeric form composed of ten or more subunits [26]. Additionally, some other dissociated forms of CRP have been reported, such as dimers, trimers, tetramers, and even other non-native pentameric configurations that have been formed again due to alterations of the microenvironment [27,28]. Apart from the pentameric ring-like form that was discovered mostly on ligand-containing membranes in a calcium-dependent manner, a study on the combination of size-exclusion chromatography and electron microscopy revealed the small globulin-like form and the fibril-like structures [29]. It was suggested that the CRP can switch between these various forms under certain conditions, and this fact serves as evidence for the structural basis of multiple functions of CRP [29].
Moreover, even though CRP was known to be a non-glycosylated protein, differentially glycosylated forms of CRP have been reported in various pathological conditions [28]. The structural integrity of CRP can also be altered because of biotinylation and denaturation [30]. Generally, several post-translational CRP modifications may lead to different protein stability and structure. Laboratory research on CRP has also revealed new forms, and the pentameric protein was found to express neo-CRP antigenicity upon various treatments, resulting in different microenvironments, but these data suggest that ligands—especially phosphocholine and antibodies—are not enough to induce neoantigenic expressions [31]. CRP multimers have been reported in vitro, along with pentamers, and it was estimated that their concentration would increase after the removal of calcium ions [32]. Crystallographic research on calcium-depleted CRP has attributed the decamer to interactions between two CRP A faces of two independent pentamers [33]. The native pentamer along with the modified monomer form are estimated to prevail; thus, this text focuses on these two forms.
2.2. Structure of C-Reactive Protein
2.2.1. The Monomeric or “Modified” CRP
X-ray crystallography has revealed that each monomer is a non-glycosylated globular subunit of 206 amino acid residues and has a molecular weight of ~23 kDa (minimum 20,946 kDa) [28,34]. It has an isoelectric point of 5.4 in contrast to the pentamer, which has an isoelectric point of 6.4. The monomer is folded into two antiparallel β-sheets with a flattened jelly roll topology similar to lectins, especially concanavalin, as well as a recognition face with a phosphocholine binding site that consists of two coordinated calcium ions adjacent to a hydrophobic pocket [35,36]. The calcium ions are bound 4 Å apart by protein sidechains deriving from long loops collected at the concave face, designated as face B, of that sheet, which is the area of ligand binding [37]. The -NH2 terminal residue of CRP is pyrrolidonecarboxylic acid, while the -COOH terminus is Pro. Furthermore, cysteine residues that form the intrachain disulfide bond are 61 residues apart in CRP primary sequence (36 and 97 residues) [34,36].
The other site is designated as face A, and carries a single α helix, thus the pentameric disc shows five helices on one face and ten calcium ions on the other [36]. Additionally, each subunit is rotated by 22° toward the fivefold axis in a way that the helices of face A are 5 Å closer to the axis, while the calcium sites of face B move out by an equivalent amount [36]. The A face includes also includes a furrow accentuated by CRP because of the substitution of few smaller sidechains and by the reorientation of some others, and defines a region that is 24 Å long, 7.5 Å deep and 12.4 Å wide [34]. The side walls consist of Ser5, Arg6, Gln203, Pro206, Trp187, Arg188, Asn160, Gly177, Leu176, Tyr175, His95, and Asp112, whereas the bottom is lined with Asn158, His38, Leu37, Val94, and Asp112 [34]. The furrows follow the monomers’ curvature and edge together closely as they enter the central pentameric pore. Also, the furrow’s outer part is positively charged, but its inner part terminates halfway through the pore at residue Asp112, resulting in a negatively charged ring lining the pore [34]. Mutagenesis research has revealed Asp112 to be a crucial residue for the recognition of C1q by CRP [38].
CRP is a calcium-dependent protein; regarding the calcium-binding cites of CRP, the first region includes Asp60, Asn61, Glu138, Asp140, and the main chain carbonyl oxygen of residue 139, yet Asp60 provides only one oxygen to the calcium ion (total of five), whereas the second equivalent cite residues contain Gln138, Asp140, and Gln150 [34]. Other data from CRP synthetic peptides show a direct binding of these two ions to a specific peptide of residues 134–148 [28]. When both calcium sites are vacant in CRP, residues 140–150 form a large loop away from the body of the molecule, exposing an otherwise hidden site of proteolysis [34]. X-ray crystallography has revealed also that these calcium ions are coordinated by Asp60, Asn61, and by residues Glu138, Gln139, Asp140, Glu147, and Gln150 in the loop; on the contrary, the past primary literature data suggest that in the first structure of CRP, the sidechain of Glu147 is not positioned to coordinate the calcium ion [28,39,40].
Primary difference maps calculated from reflection data sets accumulated from the crystals grown in the existence of phosphocholine revealed very good density for one phosphocholine molecule in each of the five CRP monomers, while the principal interaction takes place between the phosphate group of phosphocholine and the bound calcium ions [34]. Two oxygens interact directly with each calcium, leading the third oxygen away from the binding site in vitro. This orientation allows for CRP and phosphocholine interactions when the phosphate moiety is in ester linkage with other molecules, whereas the remaining phosphocholine part extends from this area and runs along the CRP surface, which is packed against Phe66, approaching the sidechain of residue Glu81 [34]. The interval between the positively charged quaternary nitrogen of phosphocholine and the acidic sidechain of Glu81 is 3.8 Å, indicating that this interaction is a critical determinant of phosphocholine binding [34]. Phe-66 and Glu-81 are the two key residues mediating the binding of phosphocholine to CRP [28]. Phe-66 accomplishes hydrophobic interactions with three methyl groups of phosphocholine, while Glu-81 is located on the opposite end of the pocket where it interacts with the nitrogen atom of choline, and the significance of both residues has been verified by mutagenesis studies [28,39]. Additionally, the Thr76 residue is a determinant of the phosphocholine-binding site as it creates the appropriately sized pocket to harbor phosphocholine [28,40]. The small sidechain of Thr76 leaves a hydrophobic cavity (8.7 × 7 × 3.5 Å) on the outer area of CRP that is lined with atoms from Glu81, Gly79, Asn61, and Thr76. This pocket encourages the creation of branched phosphocholine analogues with bulky substituents at the second position that could be bound with a higher affinity than phosphocholine [34]. Moreover, Trp67, Lys57, and Arg58 do not directly contact phosphocholine but seem to be required for the proper conformation of the binding site [40].
A small peptide at the N-terminus and another one near the C-terminus are absent in glycosylated human CRP, and their cleavage exposes two potential glycosylation sites, which are located on the opposite face from the phosphocholine-binding face of CRP [28]. In a study, the loss of these peptides exposed two possible glycosylation sites on a cleft floor, thereby keeping the protein–protein interactions in pentamers and calcium-dependent phosphocholine-binding qualitatively unaffected [41].
Furthermore, the literature data highlight that the mutagenesis of Glu42 or Pro115 due to hydrogen peroxide, which are residues in the intersubunit contact region in the pentamer, to Gln42 and Ala115, respectively, also converts CRP into biomolecules that can bind to a variety of immobilized, denatured, and aggregated proteins, thus resulting in a different final pentameric form of CRP [42]. Another study found that Thr173 and Asn186 residues are important for the binding of CRP to FcγRIIa and FcγRI [43]. Lys114, like Leu176, was found to be implicated in proteins binding to FcγRI but not FcγRIIa, whereas single mutations at amino acid positions Lys114, Asp169, Thr173, Tyr175, and Leu176 affected C1q binding to CRP, and all these results indicate a possible overlapping of these sites [43]. It is estimated that more literature data on the structure of the monomer of CRP will be evident in the near future.
2.2.2. The Pentameric or “Native” CRP
The human CRP is a pentameric member of the short pentraxin family, also known as pentraxin 1. The term “pentraxin” is derived from the Greek word for five (penta) and berry (ragos) and is related to the radial symmetry of five monomers forming a ring. It has also been used to illustrate the family of related proteins with this specific structure. Pentraxins are some highly conserved proteins—according to evolution evidence—and are supposed to precede the development of the adaptive immune response. The pentameric native form of CRP is the arrangement of five non-covalently associated monomers into a symmetric cyclic pattern around a central pore, thereby creating a discoidal and planar configuration, as seen in Figure 1.
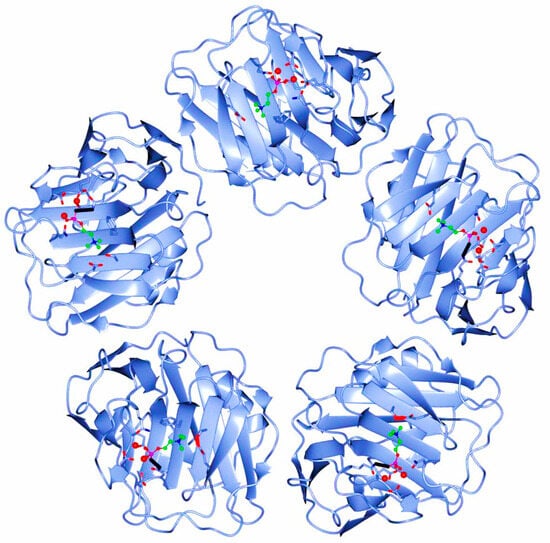
Figure 1. Pentameric structure of C-Reactive Protein (ridge helix highlighted in red).
It must be highlighted that all CRP forms are “native” as they are produced by human cells, but since the pentameric form is supposed to be the initially synthesized form, this is specifically referred to as native in current literature.
The binding of CRP to a phosphocholine-containing ligand activates the classical complement pathway up to the stage of C3 convertase, and Asp112 and Tyr175, which are residues along the boundaries of a cleft extended from each protomer’s center to the central pore of the pentamer, play critical roles in the formation of the C1q-binding site [28,34,35]. The opposite face of this pentraxin is the effector face, where complement C1q binding occurs and also Fcγ receptors are supposed to bind. A three-dimensional model for CRP with C1q binding has proposed that the acme of the predominantly positively charged C1q head domain interacts with the principally negatively charged central cavity of the CRP pentamer, and that its globular head spans the pore and interacts with two of the five protomers [44]. The strict steric requirements for this interaction imply that the ideal binding is accompanied by various slight conformational CRP changes based on each CRP ligand [44].
It was previously discussed that under certain circumstances, such as in acidic pH in vitro, CRP adopts a different pentameric configuration that exposes a hidden ligand binding site for non-phosphocholine ligands, which also enables CRP to bind to immobilized, denatured, and aggregated proteins, regardless of the identity of the native biomolecule [42]. Moreover, the literature data suggest that the fibril-like structures, which have been previously reported, are formed by the face-to-face stacking of pentamers in a number from several to hundreds, whereas the freshly purified CRPs created short single-strand fibrils that are stored for at least several days, resulting in long and bundled fibrils [29].
2.3. Genetics of C-Reactive Protein
CRP genetic locus has been mapped to the proximal long arm of chromosome 1 in the 1q23.2 region [45]. The CRP gene sequence was simultaneously discovered in 1985 by two different research teams, both reporting that it consists of 1 intron separating 2 exons [46,47]. Nucleotide sequence analysis has revealed that after coding for a signal peptide of 18 amino acids and the first two amino acids of the mature CRP, there is a long-length intron of 278 base pairs followed by the nucleotide sequence for the remaining 204 amino acids, which is the second exon, followed by a stop codon [45,47]. This unusual intron contains a poly(A) stretch that is 16 nucleotides long and a poly(GT) region that is 30 nucleotides long, which could adopt the Z-form of DNA, on the positive strand [47]. The long intron includes a GT repeat sequence, the stretch of which is polymorphic in length [45]. The mRNA cap site has been reported to be located 104 nucleotides from the beginning of the signal peptide, and there is a 3′ noncoding region with a length of 1.2 kb pairs [47]. Additionally, the gene has a typical promoter that contains the sequences TATAAAT and CAAT 29, and is 81 base pairs upstream of the cap site [47].
Despite some polymorphisms, no allelic variations or other genetic deficiencies are identified for the CRP gene. Individuals with specific allele combinations have two-fold lower baseline CRP levels, possibly due to subsequent DNA structural changes that have an impact on transcription [48]. Single Nucleotide Polymorphisms (SNPs) across the CRP gene have highlighted a significant variation in CRP levels among CRP-divergent haplotypes. CRP has also shown both decreased and/or elevated levels in various promoters [49]. Within the promoter, multiple polymorphisms have been identified in transcription factor binding E-box sites, all of which have resulted in various baseline circulating CRP titers and responses by other genes that encode cytokines that influence its synthesis, such as IL-6, IL-1, and TNF-α [45]. A systematic resequencing of the CRP gene showed as many as 40 SNPs, resulting in as many as 29 different haplotypes, with by far the highest nucleotide variance observed in African Americans, thus highlighting that the CRP gene is polymorphic [50]. Generally, multifarious CRP genetic polymorphisms have been identified in different genetic loci, which can alter CRP blood concentrations, including common CRP or new variants as well as promoter polymorphisms; these variants have been associated with an increased risk for lung cancer, coronary heart disease, and other conditions [51,52,53,54,55,56]. Nevertheless, such studies establishing associations between genetic variants and a disease risk need to be re-evaluated since potential direct molecular changes in CRP functions after genetic alterations have not yet been precisely recorded. Moreover, CRP genetic polymorphisms can affect other nearby genes; in humans, the serum amyloid P component gene and CRP gene map to 1q23.2 within an interval linked to Systemic Lupus Eruthrematosis (SLE) as well as a polymorphism related to decreased basal CRP, was also associated with the development of SLE [57].
The induction of CRP in hepatocytes is initially regulated at the transcriptional level by the cytokine Interleukin-6 (IL-6), and this effect can be enhanced by Interleukin-1β (IL-1β) since IL-6 is not sufficient by itself [58]. Although some promoter haplotypes have been associated with elevated CRP levels, this association is not IL-6-dependent, but rather reflects a change in basal promoter activity [50]. IL-6 and IL-1β regulate thew expression of several acute phase protein genes via the activation of the transcription factors STAT3, C/EBP family members, and Rel proteins belonging to NF-κB family [50,58]. The regulation of every acute phase gene is unique because of the cytokine-induced and -determined interactions of these and other transcription factors with their promoters. As a result, STAT3 is the major factor for fibrinogen genes, NF-κB is essential for the serum amyloid A gene; for CRP, the C/EBP family members C/EBPβ and C/EBPδ are crucial for induction [39]. It is important to mention that CRP and serum amyloid A share crucial amino acids, with the second one selectively modulating platelet reactivity and also down-regulating at least one CRP biological capacity. In addition to C/EBP binding sites, the direct promoter region of the CRP gene includes binding sites for STAT3 and Rel proteins [39]. Interactions between such factors that result in the enhanced stable DNA binding of C/EBP family members cause the maximum induction of the gene [39]. Additionally, transcription is regulated through E-box elements that bind the promoter to USF1, and such elements’ SNPs affect CRP levels. It is critical to note that in vitro studies on the regulation of CRP gene expression have mostly focused on primary hepatocytes, hepatocyte cell lines, or various transfected cell lines; thus, the extrahepatic production of the protein, which can show different gene expressions, has not been thoroughly studied yet [45].
This entry is adapted from the peer-reviewed paper 10.3390/diseases11040132
This entry is offline, you can click here to edit this entry!