Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
The substance P (SP)/neurokinin-1 receptor (NK-1R) system is involved in cancer progression. NK-1R, activated by SP, promotes tumor cell proliferation and migration, angiogenesis, the Warburg effect, and the prevention of apoptosis. Tumor cells overexpress NK-1R, which influences their viability. A typical specific anticancer strategy using NK-1R antagonists, irrespective of the tumor type, is possible because these antagonists block all the effects mentioned above mediated by SP on cancer cells.
- NK-1 receptor antagonist
- antitumor
- anticancer drugs
- cancer
- Aprepitant
- substance P
- repurposing
1. Introduction
Many studies have demonstrated that peptides such as neurotensin [1], angiotensin II [2], calcitonin gene-related peptide [3], somatostatin [4], and hypocretin 1 [5] are involved in cancer development. In recent years, the knowledge of the involvement of the substance P (SP)/neurokinin-1 receptor (NK-1R) system in cancer progression has notably increased [6,7,8,9,10]. It is currently known that tumor cells express receptors for peptides, explicitly emphasizing the overexpression of NK-1R reported in these cells [11]. This finding opens the door to new cancer research avenues, cancer diagnosis, and antitumor therapeutic strategies using NK-1R agonists/antagonists-based cancer therapy, cytotoxic peptide conjugate-based cancer therapy, or peptide-receptor radionuclide therapy [12,13,14,15,16,17,18,19,20]. Radiopharmaceuticals based on non-peptide compounds, such as the drug Aprepitant, a non-peptide NK-1R antagonist used in clinical practice as an antiemetic, have shown great potential in imaging studies, diagnosis, and treatment of tumors that overexpress NK-1R [16].
Based on previous in vitro and in vivo studies, the use of NK-1R antagonists such as Aprepitant is an encouraging anticancer strategy because these antagonists show a broad-spectrum anticancer effect against leukemia, sarcoma, glioma, neuroblastoma, retinoblastoma, osteosarcoma, hepatoblastoma, melanoma, or carcinoma, as a result of their ability to promote apoptosis in cancer cells [6,7,8,9,10,21]. NK-1R antagonists bind to NK-1R expressed in tumor cells and block all the favorable effects mediated by SP on tumor cells, such as proliferation and migration, prevention of apoptosis, and the Warburg effect (glycolytic rate increase) and angiogenesis [22].
2. Involvement of the SP/NK-1R System in Cancer
The general characteristics of the SP/NK-1R system and the primary data demonstrating its involvement in cancer progression are included in the following thirteen key points [6,7,24,25,26,27,28,29,30,31,32,33,34,35,36] (Figure 1):
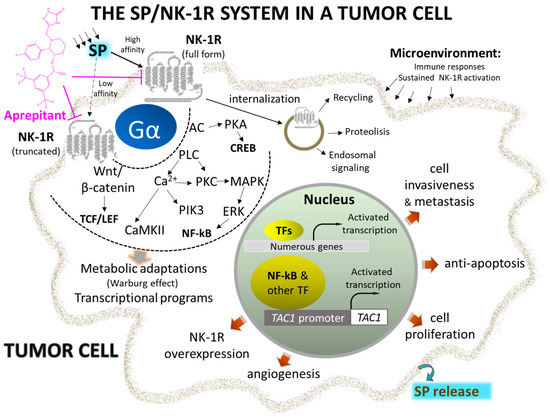
Figure 1. Representative signaling pathways and activated transcriptional programs implicating complete and truncated NK-1R isoforms in the initiation and metastasis of cancer cells. Overexpression of NK-1R and agonist overstimulation induce metabolic changes and transcriptional programs, leading to uncontrolled cell proliferation and migration. Selective and potent antagonists, such as Aprepitant, block the signal and stimulate apoptosis of cancer cells (see text for details). Abbreviations: AC, adenylyl cyclase; CaMKII, calcium-calmodulin kinase II; CREB, cAMP response element; ERK, extracellular-activated kinase; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NK-1R, neurokinin 1-receptor; PIK3, phosphatidylinositol 3 kinase; PKC, protein kinase C; PLC, phospholipase C; SP, substance P; TAC1, tachykinin precursor 1; TCF/LEF, T cell factor/lymphoid enhancer factor family of transcription factors; Wnt, wingless and Int-1.
-
NK-1R belongs to the rhodopsin-like G protein-coupled receptors family and shows a preferential affinity for SP [37]. This peptide is the natural ligand for NK-1R, so NK-1R is also named the SP receptor [38,39]. The SP high-affinity receptor NK-1R is widely distributed throughout the body and can bind to hemokinin-1, endokinins, and neurokinins. For a recent review focused on the structural dynamics and signaling of NK-1R, see [23].
-
The 5’ flanking region of the tachykinin receptor 1 gene contains conserved gene promoter regulatory elements such as the octamer binding protein 2, nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB), activating protein-1, -2, and -4, and cAMP responsive element binding sites. NF-κB favors cell survival, DNA transcription, and cancer progression, blocks apoptosis, and promotes cancer resistance and the synthesis of tumor-associated cytokines (interferon γ, macrophage inflammatory protein-1β, tumor necrosis factor α, interleukins 1β and 6) in tumor cells [42].
-
NK-1R is coupled to Gαs, Gαq, Gαo, Gαi, and Gα12/13 proteins; activation of a specific G protein, which differs in the signaling/effector pathways they activate, is controlled by the conformations of NK-1R (each conformation shows a distinct affinity for antagonists and agonists) and type of ligands [43,44,45,46,47,48,49]. Aprepitant promotes conformational changes that interfere with the binding of SP, leading to a long-lasting inhibition of NK-1R [22,50,51,52]. The interaction between SP central and N-terminal regions with the NK-1R extracellular domain is crucial for signaling through Gs. NK-1R antagonists block access to the receptor binding site and hinder G-protein activation [53].
-
SP binds to the extracellular loops of NK-1R [54] and non-peptide NK-1R antagonists between the receptor’s III and VI transmembrane segments; specific amino acid residues (His197, Gln165) control the binding of these antagonists as reported in recent detailed structural studies [48,52,53,55]. The amidated C-terminal of SP is involved in peptide activity since its deamidation suppressed the activity of SP. The C-terminal sequence contains a hydrophobic amino acid residue needed to activate NK-1R [53].
-
Cancer cells express/overexpress NK-1R [11,56,57,58,59,60,61,62,63,64], which is involved in the viability of these cells. It has been suggested that the higher the number of NK-1R, the higher the tumor malignancy, and NK-1R mRNA expression is lower in benign compared to malignant tissues. NK-1R is not involved in the viability of normal cells. Tumor cells express and release SP, but SP is not involved in the viability of cancer cells [65].
-
SP favors the proliferation of tumor cells [66,67,68,69,70,71,72]. The peptide, through NK-1R, promotes the proliferation via mitogen-activated protein kinases (MAPK) signaling and migration of both solid and non-solid cancer cells via the expression of matrix metalloproteinase 9 and exerts an antiapoptotic action by activating protein kinase B (this has been associated with a poor prognosis). SP favors the Warburg effect (glycolytic rate increase), which cancer cells use to maintain their high metabolism rate. SP promotes angiogenesis, leading to neovascularization [73]. Consequently, rapidly multiplying cancer cells use the nutrients and oxygen provided due to SP-induced angiogenesis. SP also increases the expression of NK-1R but not that of the other two tachykinin receptors (NK-2R and NK-3R).
-
NK-1R shows two isoforms: full-length (407 amino acids) and truncated (311 amino acids; C-terminus 96 residues are lost) [74,75]. Isoforms can trigger different intracellular signaling pathways and play different roles in physiological and pathophysiological mechanisms [49]. The full-length form shows a ten-fold higher binding affinity for SP than the truncated form. Cells expressing the full-length form respond to nanomolar concentrations of SP, whereas those cells expressing the truncated isoform need micromolar concentrations of the peptide to elicit a signaling response. The full-length isoform is involved in NK-1R desensitization and internalization, whereas the truncated form partially disrupts signaling pathways but does not affect the SP binding domain. Truncation of the receptor leads to impairment in the receptor being internalized, thus imparting the receptor with (a) resistance to desensitization, (b) weaker interaction with G proteins and protein kinase K and other phosphorylation processes, and (c) delay in Ca++ release ultimately resulting in an inhibited response to SP.
-
Tumor cells show a higher level of truncated and a lower level of full-length NK-1R than normal cells. MicroRNA-206 overexpression favors cancer cell proliferation, invasion, and migration via targeting the full-length form, whereas microRNA-22 blocks all these processes by targeting the truncated NK-1R form [67]. Furthermore, NK-1R is the predicted target of the miR-34 family, and the overexpression of microRNA-34b/c-5p has been shown to suppress cancer cell proliferation and promote apoptosis via NK-1R suppression.
-
The truncated NK-1R isoform is involved in malignancy, tumor cell growth, metastasis, and apoptosis blockade [76]. In contrast, the full-length expression, inversely associated with invasion and metastasis, decreases cancer cell proliferation and attenuates apoptotic signals. Truncated NK-1 expression is positively regulated via Smad4 by tumor growth factor β and blocked with NK-1R antagonists (Aprepitant). SP promotes the activation of NF-κB, which upregulates the truncated form, induces a slight increase in the full-length isoform, and favors resistance to some chemotherapeutic agents and Aprepitant. This observation is vital since tumors showing overexpression of the NF-κB pathway may need to be treated with a higher dose of the NK-1R antagonist for mediating anticancer effects.
-
SP is synthesized and released by cancer and immune cells, and it is released from nerve terminals and circulates in the bloodstream [62,77,78,79]. SP-immunoreactive fibers have been associated with tumor differentiation status. SP acts through autocrine, paracrine, neuroendocrine, and endocrine (from the tumor mass) mechanisms. SP and NK-1R have also been observed in the nuclei of cancer cells; their physiological significance is currently unknown.
-
Following the interaction of SP with NK-1R, important downstream events are activated, such as diacylglycerol synthesis with the resultant activation of protein kinase C and promotion of the influx of extracellular Ca++ via calcium channels. Downstream events, such as inositol triphosphate production, promote Ca++ release from the endoplasmic reticulum into the cytoplasm. High calcium levels enhance proliferative and pro-survival pathways such as MAPK and extracellular signal-regulated kinases (ERK). Furthermore, calcium governs other essential processes such as cell death, migration, communication, and immune activation [80]. NK-1R antagonists promote rapid endoplasmic reticulum/mitochondria Ca++ overload and accumulation of reactive oxygen species, causing apoptosis. In conclusion, since the processes mentioned above can be deregulated and exploited by cancer cells that overexpress NK-1R, this receptor can serve as a suitable therapeutic target in cancer [50,81,82].
-
Although both normal and cancer cells produce SP, lower levels of SP are detected in normal cells compared with cancer cells. More serum SP and NK-1R concentrations were found in cancer patients than healthy individuals. NK-1R overexpression has been associated with larger tumor size, higher metastatic and invasion potential tumor-node metastasis, advanced cancer stages, and poor prognosis. Thus, NK-1R overexpression and high serum SP levels could be used as predictive biomarkers for increased risk of developing cancer and cancer prognosis [57,59].
3. NK-1R Antagonists as Anticancer Drugs
Non-peptide NK-1R antagonists are lipid-soluble compounds that show a different chemical composition (benzyl ether piperidines, benzylamine/benzyl ether quinuclidine, benzyl amino piperidines, steroids, tryptophan derivatives, perhydro isoindolines) (Figure 2) but have a similar affinity for NK-1R [52,85].
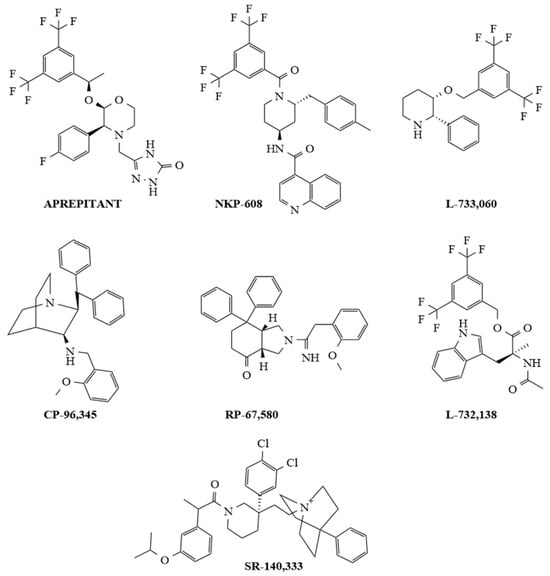
Figure 2. Chemical structures of representative selective non-peptide NK-1R antagonists.
SP promotes blood–brain barrier breaching by tumor cells, and most importantly, non-peptide NK-1R antagonists easily cross the blood–brain barrier, and peptidases do not degrade these antagonists. Additionally, non-peptide NK-1R antagonists decrease the toxicity of cytostatics and the permeability of tumor cells across this barrier (preventing brain metastasis); in general, they are safe and well tolerated [6,81,86,87]. Many in vitro and in vivo studies have demonstrated that NK-1R antagonists (Aprepitant, L-732,138, L-733,060, CP-96,345, SR-140,333, NKP-608, RP-67,580) are broad-spectrum anticancer agents that act in a concentration-dependent manner [22,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104]; one of the primary mechanisms by which these antagonists induce an antitumor effect is by causing the death of many different cancer cell types by apoptosis (see Figure 2).
Testing the anticancer action of NK-1R antagonists in other tumors, such as oral squamous cell carcinoma and uterine leiomyomata, is worthwhile since all these tumor types express NK-1R [11,129]. It is important to note that the antiemetic drug Aprepitant is the NK-1R antagonist exerting an anticancer action (apoptosis) against as many as 21 different tumor types; this is pivotal for its repurposing as an anticancer drug. As stated above, SP impairs the blood–brain barrier, thus facilitating the invasion of cancer cells into the central nervous system; NK-1R antagonists have been shown to prevent this invasion by cancer cells [86]. The SP/NK-1R interaction pathway induces the migration of tumor cells via Rho-associated protein kinase (ROCK)-mediated signaling, leading to the upregulation of expression of matrix metalloproteinase 2, which degrades extracellular matrix proteins [41,130,131]. NK-1R antagonists are known to prevent tumor cell proliferation, invasion, and metastasis via suppression of the Wnt/β-catenin signaling pathway, and it has been suggested that, before and after cancer surgical procedures, Aprepitant could be administered to prevent metastasis and recurrence [132,133]. Cancer cells showing high levels of the truncated NK-1R form are highly responsive to NK-1R antagonists, and this could be important for the specific and safe use of NK-1R antagonists since tumor cells express more of this form than normal cells [103]
Moreover, NK-1R agonists decreased the expression of Dickkopf 1 (a Wnt inhibitor) and augmented β-catenin and glycogen synthase kinase-3β expressions [134]. The NK-1R antagonist Aprepitant favors caspase-dependent apoptotic mechanisms. It alters the expression of genes involved in cell survival and drug resistance by blocking the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling cascade. It increases the cleavage of the poly (ADP-ribose) polymerase, an enzyme that repairs DNA damage [95,97,135]. Aprepitant shows mild/moderate side effects: the most common are constipation, diarrhea, headache, hiccups, fatigue, and anorexia; however, other low-incidence side effects (<1%) such as euphoria, disorientation, cognitive disorders, candidiasis, acid reflux, epigastric discomfort, and lethargy have also been reported [81,136,137]. Aprepitant is currently used as an antiemetic but also exerts an antipruritic action in cutaneous lymphoma and acts as a cough suppressant in patients with lung cancer [6,138,139]. Because Aprepitant is poorly water-soluble, strategies (e.g., nanoparticle formulation) must be developed to increase its solubility, dissolution, and efficacy [6]. Finally, a liquid chromatography–tandem mass spectrometry method has recently been developed to quantify the free and total Aprepitant and its active N-dealkylated metabolites in human plasma, allowing accurate measurement in pharmacokinetic studies [140].
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115936
This entry is offline, you can click here to edit this entry!