Capsaicinoids are a unique chemical species resulting from a particular biosynthesis pathway of hot chilies (Capsicum spp.) that gives rise to 22 analogous compounds, all of which are TRPV1 agonists and, therefore, responsible for the pungency of Capsicum fruits. In addition to their human consumption, numerous ethnopharmacological uses of chili have emerged throughout history. Today, more than 25 years of basic research accredit a multifaceted bioactivity mainly to capsaicin, highlighting its antitumor properties mediated by cytotoxicity and immunological adjuvancy against at least 74 varieties of cancer, while non-cancer cells tend to have greater tolerance.
- apoptosis
- autophagy
- vanilloid-like transient potential receptors (TRPV)
- tumor-associated NADH oxidase (tNOX)
- reactive oxygen species (ROS)
- silent mating-type information regulation 2 homolog 1 (SIRT1)
- p53
1. Introduction
2. Molecular Mechanisms behind the Selective Cytotoxicity of Capsaicin in Cancer Cells
3. Mechanisms Underlying Capsaicin-Induced Apoptosis in Cancer: Brief State of the Art
3.1. Contribution of Oxidative Stress in the Proapoptotic Activity of Capsaicin in Cancer
3.2. Extrinsic Apoptosis Events Involved in the Cytotoxic Activity of Capsaicin in Cancer
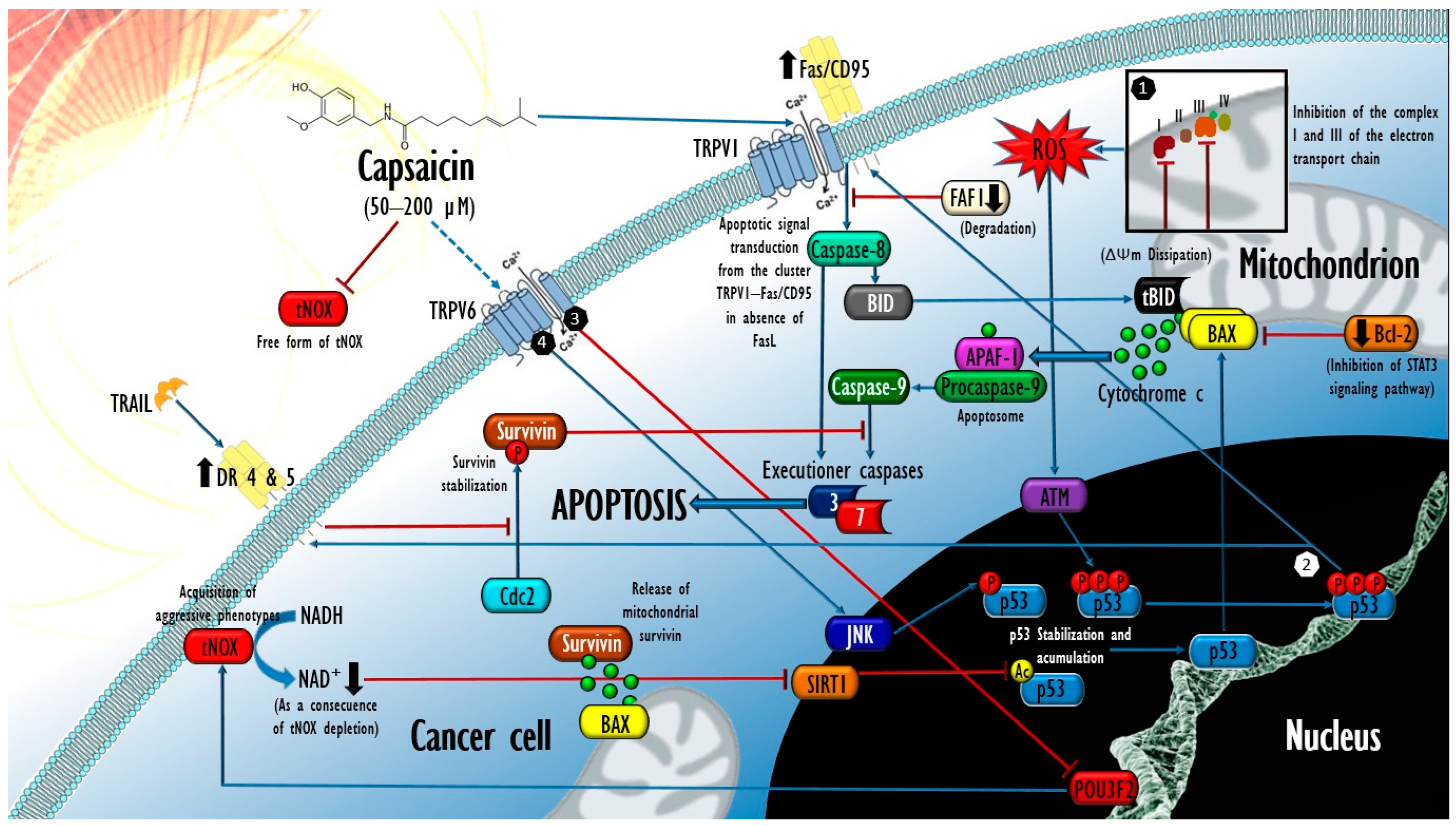
3.3. In Vivo Antitumor Activity of Capsaicin and Expectations for Its Clinical Evaluation
This entry is adapted from the peer-reviewed paper 10.3390/cells12212573
References
- Adetunji, T.L.; Olawale, F.; Olisah, C.; Adetunji, A.E.; Aremu, A.O. Capsaicin: A Two-Decade Systematic Review of Global Research Output and Recent Advances Against Human Cancer. Front. Oncol. 2022, 12, 908487.
- Kim, S.; Park, M.; Yeom, S.I.; Kim, Y.M.; Lee, J.M.; Lee, H.A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.T.; et al. Genome Sequence of the Hot Pepper Provides Insights into the Evolution of Pungency in Capsicum Species. Nat. Genet. 2014, 46, 270–278.
- Cordell, G.A.; Araujo, O.E. Capsaicin: Identification, Nomenclature, and Pharmacotherapy. Ann. Pharmacother. 1993, 27, 330–336.
- Contreras-Padilla, M.; Yahia, E.M. Changes in Capsaicinoids during Development, Maturation, and Senescence of Chile Peppers and Relation with Peroxidase Activity. J. Agric. Food Chem. 1998, 46, 2075–2079.
- Chung, M.K.; Güler, A.D.; Caterina, M.J. TRPV1 Shows Dynamic Ionic Selectivity during Agonist Stimulation. Nat. Neurosci. 2008, 11, 555–564.
- Perry, L.; Dickau, R.; Zarrillo, S.; Holst, I.; Pearsall, D.M.; Piperno, D.R.; Berman, M.J.; Cooke, R.G.; Rademaker, K.; Ranere, A.J.; et al. Starch Fossils and the Domestication and Dispersal of Chili Peppers (Capsicum spp. L.) in the Americas. Science 2007, 315, 986–988.
- Halikowski Smith, S. In the Shadow of a Pepper-Centric Historiography: Understanding the Global Diffusion of Capsicums in the Sixteenth and Seventeenth Centuries. J. Ethnopharmacol. 2015, 167, 64–77.
- Cichewicz, R.H.; Thorpe, P.A. The Antimicrobial Properties of Chile Peppers (Capsicum species) and Their Uses in Mayan Medicine. J. Ethnopharmacol. 1996, 52, 61–70.
- Meghvansi, M.K.; Siddiqui, S.; Khan, M.H.; Gupta, V.K.; Vairale, M.G.; Gogoi, H.K.; Singh, L. Naga Chilli: A Potential Source of Capsaicinoids with Broad-Spectrum Ethnopharmacological Applications. J. Ethnopharmacol. 2010, 132, 1–14.
- Materska, M.; Konopacka, M.; Rogoliński, J.; Ślosarek, K. Antioxidant Activity and Protective Effects against Oxidative Damage of Human Cells Induced by X-Radiation of Phenolic Glycosides Isolated from Pepper Fruits Capsicum annuum L. Food Chem. 2015, 168, 546–553.
- Wahyuni, Y.; Ballester, A.R.; Sudarmonowati, E.; Bino, R.J.; Bovy, A.G. Secondary Metabolites of Capsicum Species and Their Importance in the Human Diet. J. Nat. Prod. 2013, 76, 783–793.
- Wahyuni, Y.; Ballester, A.R.; Sudarmonowati, E.; Bino, R.J.; Bovy, A.G. Metabolite Biodiversity in Pepper (Capsicum) Fruits of Thirty-Two Diverse Accessions: Variation in Health-Related Compounds and Implications for Breeding. Phytochemistry 2011, 72, 1358–1370.
- Tundis, R.; Menichini, F.; Bonesi, M.; Conforti, F.; Statti, G.; Menichini, F.; Loizzo, M.R. Antioxidant and Hypoglycaemic Activities and Their Relationship to Phytochemicals in Capsicum annuum Cultivars during Fruit Development. LWT 2013, 53, 370–377.
- Schwarzlin, R.; Pušenjak, N.; Makuc, D.; Križman, M.; Vovk, I.; Plavec, J.; Švajger, U. Synergistic Complex from Plants Solanaceae Exhibits Cytotoxicity for the Human Hepatocellular Carcinoma Cell Line HepG2. BMC Complement. Altern. Med. 2016, 16, 395.
- Zimmer, A.R.; Leonardi, B.; Miron, D.; Schapoval, E.; De Oliveira, J.R.; Gosmann, G. Antioxidant and Anti-Inflammatory Properties of Capsicum baccatum: From Traditional Use to Scientific Approach. J. Ethnopharmacol. 2012, 139, 228–233.
- Sricharoen, P.; Lamaiphan, N.; Patthawaro, P.; Limchoowong, N.; Techawongstien, S.; Chanthai, S. Phytochemicals in Capsicum Oleoresin from Different Varieties of Hot Chilli Peppers with Their Antidiabetic and Antioxidant Activities Due to Some Phenolic Compounds. Ultrason. Sonochem. 2017, 38, 629–639.
- Fernández-Bedmar, Z.; Alonso-Moraga, A. In Vivo and in Vitro Evaluation for Nutraceutical Purposes of Capsaicin, Capsanthin, Lutein and Four Pepper Varieties. Food Chem. Toxicol. 2016, 98, 89–99.
- Alonso-Castro, A.J.; Domínguez, F.; Zapata-Morales, J.R.; Carranza-Álvarez, C. Plants Used in the Traditional Medicine of Mesoamerica (Mexico and Central America) and the Caribbean for the Treatment of Obesity. J. Ethnopharmacol. 2015, 175, 335–345.
- Majee, S.K.; Ray, S.; Ghosh, K.; Micard, V.; Ray, B. Isolation and Structural Features of an Antiradical Polysaccharide of Capsicum annuum That Interacts with BSA. Int. J. Biol. Macromol. 2015, 75, 144–151.
- Sung, J.; Jeong, H.S.; Lee, J. Effect of the Capsicoside G-Rich Fraction from Pepper (Capsicum annuum L.) Seeds on High-Fat Diet-Induced Obesity in Mice. Phytother. Res. 2016, 30, 1848–1855.
- Allemand, A.; Leonardi, B.F.; Zimmer, A.R.; Moreno, S.; Romão, P.R.T.; Gosmann, G. Red Pepper (Capsicum baccatum) Extracts Present Anti-Inflammatory Effects In Vivo and Inhibit the Production of TNF-α and NO In Vitro. J. Med. Food 2016, 19, 759–767.
- Brito-Argáez, L.; Tamayo-Sansores, J.A.; Madera-Piña, D.; García-Villalobos, F.J.; Moo-Puc, R.E.; Kú-González, Á.; Villanueva, M.A.; Islas-Flores, I. Biochemical Characterization and Immunolocalization Studies of a Capsicum Chinense Jacq. Protein Fraction Containing DING Proteins and Anti-Microbial Activity. Plant Physiol. Biochem. 2016, 109, 502–514.
- Lv, J.; Qi, L.; Yu, C.; Yang, L.; Guo, Y.; Chen, Y.; Bian, Z.; Sun, D.; Du, J.; Ge, P.; et al. Consumption of Spicy Foods and Total and Cause Specific Mortality: Population Based Cohort Study. BMJ 2015, 351, h3942.
- Chopan, M.; Littenberg, B. The Association of Hot Red Chili Pepper Consumption and Mortality: A Large Population-Based Cohort Study. PLoS ONE 2017, 12, e0169876.
- Xue, Y.; He, T.; Yu, K.; Zhao, A.; Zheng, W.; Zhang, Y.; Zhu, B. Association between Spicy Food Consumption and Lipid Profiles in Adults: A Nationwide Population-Based Study. Br. J. Nutr. 2017, 118, 144–153.
- Asnin, L.; Park, S.W. Isolation and Analysis of Bioactive Compounds in Capsicum Peppers. Crit. Rev. Food Sci. Nutr. 2015, 55, 254–289.
- Sharma, S.K.; Vij, A.S.; Sharma, M. Mechanisms and Clinical Uses of Capsaicin. Eur. J. Pharmacol. 2013, 720, 55–62.
- Abdel Salam, O. Capsaicin as a Therapeutic Molecule; Springer: Basel, Switzerland, 2014; Volume 68, p. 321.
- Srinivasan, K. Biological Activities of Red Pepper (Capsicum annuum) and Its Pungent Principle Capsaicin: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1488–1500.
- Basith, S.; Cui, M.; Hong, S.; Choi, S. Harnessing the Therapeutic Potential of Capsaicin and Its Analogues in Pain and Other Diseases. Molecules 2016, 21, 966.
- Fattori, V.; Hohmann, M.S.N.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Verri, W.A. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules 2016, 21, 844.
- Akhtar, F.; Sharif, H.; Mallick, M.; Zahoor, F.; Abdulmalik, A.; Baig, W.; Shujaat, N.; Gul, S.; Bibi, G.; Ramzan, R.; et al. Capsaicin: Its biological activities and in silico target fishing. Acta Pol. Pharm. 2017, 74, 321–329.
- Adaszek, Ł.; Gadomska, D.; Mazurek, Ł.; Łyp, P.; Madany, J.; Winiarczyk, S. Properties of Capsaicin and Its Utility in Veterinary and Human Medicine. Res. Vet. Sci. 2019, 123, 14–19.
- Sanati, S.; Razavi, B.M.; Hosseinzadeh, H. A Review of the Effects of Capsicum annuum L. And Its Constituent, Capsaicin, in Metabolic Syndrome. Iran J. Basic Med. Sci. 2018, 21, 439–448.
- Galati, G.; O’brien, P.J. Cytoprotective and Anticancer Properties of Coenzyme Q versus Capsaicin; IOS Press: Amsterdam, The Netherlands, 2003; Volume 18.
- Impheng, H.; Pongcharoen, S.; Richert, L.; Pekthong, D.; Srisawang, P. The Selective Target of Capsaicin on FASN Expression and de Novo Fatty Acid Synthesis Mediated through ROS Generation Triggers Apoptosis in HepG2 Cells. PLoS ONE 2014, 9, e107842.
- Kim, J.Y.; Kim, E.H.; Kim, S.U.; Kwon, T.K.; Choi, K.S. Capsaicin Sensitizes Malignant Glioma Cells to TRAIL-Mediated Apoptosis via DR5 Upregulation and Survivin Downregulation. Carcinogenesis 2010, 31, 367–375.
- Lau, J.K.; Brown, K.C.; Dom, A.M.; Witte, T.R.; Thornhill, B.A.; Crabtree, C.M.; Perry, H.E.; Brown, J.M.; Ball, J.G.; Creel, R.G.; et al. Capsaicin Induces Apoptosis in Human Small Cell Lung Cancer via the TRPV6 Receptor and the Calpain Pathway. Apoptosis 2014, 19, 1190–1201.
- Wang, P.; Sun, Y.C.; Lu, W.H.; Huang, P.; Hu, Y. Selective Killing of K-Ras–Transformed Pancreatic Cancer Cells by Targeting NAD(P)H Oxidase. Chin. J. Cancer 2015, 34, 1–11.
- Lee, Y.H.; Chen, H.Y.; Su, L.J.; Chueh, P.J. Sirtuin 1 (SIRT1) Deacetylase Activity and NAD+/NADH Ratio Are Imperative for Capsaicin-Mediated Programmed Cell Death. J. Agric. Food Chem. 2015, 63, 7361–7370.
- Ghosh, A.K.; Basu, S. Fas-Associated Factor 1 Is a Negative Regulator in Capsaicin Induced Cancer Cell Apoptosis. Cancer Lett. 2010, 287, 142–149.
- Beltran, J.; Ghosh, A.K.; Basu, S. Immunotherapy of Tumors with Neuroimmune Ligand Capsaicin. J. Immunol. 2007, 178, 3260–3264.
- Macho, A.; Calzado, M.A.; Muñoz-Blanco, J.; Gómez-Díaz, C.; Gajate, C.; Mollinedo, F.; Navas, P.; Muñoz, E. Selective Induction of Apoptosis by Capsaicin in Transformed Cells: The Role of Reactive Oxygen Species and Calcium. Cell Death Differ. 1999, 6, 155–165.
- Bley, K.; Boorman, G.; Mohammad, B.; McKenzie, D.; Babbar, S. A Comprehensive Review of the Carcinogenic and Anticarcinogenic Potential of Capsaicin. Toxicol. Pathol. 2012, 40, 847–873.
- Pramanik, K.C.; Srivastava, S.K. Role of Capsaicin in Cancer Prevention. In Role Capsaicin Oxidative Stress Cancer; Springer: Dordrecht, The Netherlands, 2013; pp. 1–18.
- Clark, R.; Lee, S.-H. Anticancer Properties of Capsaicin Against Human Cancer. Anticancer Res. 2016, 36, 837–843.
- Kim, C.S.; Park, W.H.; Park, J.Y.; Kang, J.H.; Kim, M.O.; Kawada, T.; Yoo, H.; Han, I.S.; Yu, R. Capsaicin, a Spicy Component of Hot Pepper, Induces Apoptosis by Activation of the Peroxisome Proliferator-Activated Receptor Gamma in HT-29 Human Colon Cancer Cells. J. Med. Food 2004, 7, 267–273.
- Qiao, S.; Li, W.; Tsubouchi, R.; Haneda, M.; Murakami, K.; Yoshino, M. Involvement of Peroxynitrite in Capsaicin-Induced Apoptosis of C6 Glioma Cells. Neurosci. Res. 2005, 51, 175–183.
- Sánchez, A.M.; Sánchez, M.G.; Malagarie-Cazenave, S.; Olea, N.; Díaz-Laviada, I. Induction of Apoptosis in Prostate Tumor PC-3 Cells and Inhibition of Xenograft Prostate Tumor Growth by the Vanilloid Capsaicin. Apoptosis 2006, 11, 89–99.
- Athanasiou, A.; Smith, P.A.; Vakilpour, S.; Kumaran, N.M.; Turner, A.E.; Bagiokou, D.; Layfield, R.; Ray, D.E.; Westwell, A.D.; Alexander, S.P.H.; et al. Vanilloid Receptor Agonists and Antagonists Are Mitochondrial Inhibitors: How Vanilloids Cause Non-Vanilloid Receptor Mediated Cell Death. Biochem. Biophys. Res. Commun. 2007, 354, 50–55.
- Wang, S.; Morré, D.M.; Morré, D.J. Sera from Cancer Patients Contain Two Oscillating ECTO-NOX Activities with Different Period Lengths. Cancer Lett. 2003, 190, 135–141.
- Cho, N.; Morré, D.J. Early Developmental Expression of a Normally Tumor-Associated and Drug-Inhibited Cell Surface-Located NADH Oxidase (ENOX2) in Non-Cancer Cells. Cancer Immunol. Immunother. 2009, 58, 547–552.
- Morré, D.J.; Morré, D.M. ECTO-NOX Proteins: Growth, Cancer, and Aging; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; pp. 1–507.
- Morré, D.J.; Hostetler, B.; Weston, N.; Kim, C.; Morré, D.M. Cancer Type-Specific TNOX Isoforms: A Putative Family of Redox Protein Splice Variants with Cancer Diagnostic and Prognostic Potential. Biofactors 2008, 34, 201–207.
- Cheng, H.L.; Lee, Y.H.; Yuan, T.M.; Chen, S.W.; Chueh, P.J. Update on a Tumor-Associated NADH Oxidase in Gastric Cancer Cell Growth. World J. Gastroenterol. 2016, 22, 2900–2905.
- Morré, D.J. NADH Oxidase: A Multifunctional Ectoprotein of the Eukaryotic Cell Surface. In Plasma Membrane Redox Systems and Their Role in Biological Stress and Disease; Springer: Dordrecht, The Netherlands, 1998; pp. 121–156.
- Liu, S.C.; Yang, J.J.; Shao, K.N.; Chueh, P.J. RNA Interference Targeting TNOX Attenuates Cell Migration via a Mechanism That Involves Membrane Association of Rac. Biochem. Biophys. Res. Commun. 2008, 365, 672–677.
- Liu, N.C.; Hsieh, P.F.; Hsieh, M.K.; Zeng, Z.M.; Cheng, H.L.; Liao, J.W.; Chueh, P.J. Capsaicin-Mediated TNOX (ENOX2) up-Regulation Enhances Cell Proliferation and Migration In Vitro and In Vivo. J. Agric. Food Chem. 2012, 60, 2758–2765.
- Zeng, Z.M.; Chuang, S.M.; Chang, T.C.; Hong, C.W.; Chou, J.C.; Yang, J.J.; Chueh, P.J. Phosphorylation of Serine-504 of TNOX (ENOX2) Modulates Cell Proliferation and Migration in Cancer Cells. Exp. Cell Res. 2012, 318, 1759–1766.
- Islam, A.; Su, A.J.; Zeng, Z.M.; Chueh, P.J.; Lin, M.H. Capsaicin Targets TNOX (ENOX2) to Inhibit G1 Cyclin/CDK Complex, as Assessed by the Cellular Thermal Shift Assay (CETSA). Cells 2019, 8, 1275.
- Morré, D.J.; Caldwell, S.; Mayorga, A.; Wu, L.Y.; Morré, D.M. NADH Oxidase Activity from Sera Altered by Capsaicin Is Widely Distributed among Cancer Patients. Arch. Biochem. Biophys. 1997, 342, 224–230.
- Morré, D.J.; Bridge, A.; Wu, L.Y.; Morré, D.M. Preferential Inhibition by (-)-Epigallocatechin-3-Gallate of the Cell Surface NADH Oxidase and Growth of Transformed Cells in Culture. Biochem. Pharmacol. 2000, 60, 937–946.
- Chueh, P.J.; Kim, C.; Cho, N.M.; Morré, D.M.; Morré, D.J. Molecular Cloning and Characterization of a Tumor-Associated, Growth-Related, and Time-Keeping Hydroquinone (NADH) Oxidase (TNOX) of the HeLa Cell Surface. Biochemistry 2002, 41, 3732–3741.
- Chueh, P.-J.; Wu, L.-Y.; Morré, D.M.; Morré, D.J. TNOX Is Both Necessary and Sufficient as a Cellular Target for the Anticancer Actions of Capsaicin and the Green Tea Catechin (-)-Epigallocatechin-3-Gallate. Biofactors 2004, 20, 235–249.
- Wang, H.M.; Chueh, P.J.; Chang, S.P.; Yang, C.L.; Shao, K.N. Effect of Ccapsaicin on TNOX (ENOX2) Protein Expression in Stomach Cancer Cells. Biofactors 2008, 34, 209–217.
- Lin, M.H.; Lee, Y.H.; Cheng, H.L.; Chen, H.Y.; Jhuang, F.H.; Chueh, P.J. Capsaicin Inhibits Multiple Bladder Cancer Cell Phenotypes by Inhibiting Tumor-Associated NADH Oxidase (TNOX) and Sirtuin1 (SIRT1). Molecules 2016, 21, 849.
- Wang, H.M.; Chuang, S.M.; Su, Y.C.; Li, Y.H.; Chueh, P.J. Down-Regulation of Tumor-Associated NADH Oxidase, TNOX (ENOX2), Enhances Capsaicin-Induced Inhibition of Gastric Cancer Cell Growth. Cell Biochem. Biophys. 2011, 61, 355–366.
- Chang, C.-F.; Islam, A.; Liu, P.-F.; Zhan, J.-H.; Chueh, J. Capsaicin Acts through TNOX (ENOX2) to Induce Autophagic Apoptosis in P53-Mutated HSC-3 Cells but Autophagy in P53-Functional SAS Oral Cancer Cells. Am. J. Cancer Res. 2020, 10, 3230.
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and Paxillin Dynamics at Focal Adhesions in the Protrusions of Migrating Cells. Sci. Rep. 2014, 4, srep06024.
- Pramanik, K.C.; Fofaria, N.M.; Gupta, P.; Srivastava, S.K. CBP-Mediated FOXO-1 Acetylation Inhibits Pancreatic Tumor Growth by Targeting SirT. Mol. Cancer Ther. 2014, 13, 687–698.
- Chow, J.; Norng, M.; Zhang, J.; Chai, J. TRPV6 Mediates Capsaicin-Induced Apoptosis in Gastric Cancer Cells--Mechanisms behind a Possible New “Hot” Cancer Treatment. Biochim. Biophys. Acta 2007, 1773, 565–576.
- Skrzypski, M.; Sassek, M.; Abdelmessih, S.; Mergler, S.; Grötzinger, C.; Metzke, D.; Wojciechowicz, T.; Nowak, K.W.; Strowski, M.Z. Capsaicin Induces Cytotoxicity in Pancreatic Neuroendocrine Tumor Cells via Mitochondrial Action. Cell Signal. 2014, 26, 41–48.
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of Vanilloid Transient Receptor Potential Cation Channels. Mol. Pharmacol. 2009, 75, 1262–1279.
- Hoenderop, J.G.J.; Hartog, A.; Stuiver, M.; Doucet, A.; Willems, P.H.G.M.; Bindels, R.J.M. Localization of the Epithelial Ca(2+) Channel in Rabbit Kidney and Intestine. J. Am. Soc. Nephrol. 2000, 11, 1171–1178.
- Roderick, H.L.; Cook, S.J. Ca2+ Signalling Checkpoints in Cancer: Remodelling Ca2+ for Cancer Cell Proliferation and Survival. Nat. Rev. Cancer 2008, 8, 361–375.
- van Abel, M.; Hoenderop, J.G.J.; Bindels, R.J.M. The Epithelial Calcium Channels TRPV5 and TRPV6: Regulation and Implications for Disease. Naunyn. Schmiedebergs Arch. Pharmacol. 2005, 371, 295–306.
- Woudenberg-Vrenken, T.E.; Lameris, A.L.; Weißgerber, P.; Olausson, J.; Flockerzi, V.; Bindels, R.J.M.; Freichel, M.; Hoenderop, J.G.J. Functional TRPV6 Channels Are Crucial for Transepithelial Ca2+ Absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G879–G885.
- Seebohm, G.; Schreiber, J.A. Beyond Hot and Spicy: TRPV Channels and Their Pharmacological Modulation. Cell Physiol. Biochem. 2021, 22, 108–130.
- Peng, J.B.; Zhuang, L.; Berger, U.V.; Adam, R.M.; Williams, B.J.; Brown, E.M.; Hediger, M.A.; Freeman, M.R. CaT1 Expression Correlates with Tumor Grade in Prostate Cancer. Biochem. Biophys. Res. Commun. 2001, 282, 729–734.
- Wissenbach, U.; Niemeyer, B.A.; Fixemer, T.; Schneidewind, A.; Trost, C.; Cavalié, A.; Reus, K.; Meese, E.; Bonkhoff, H.; Flockerzi, V. Expression of CaT-like, a Novel Calcium-Selective Channel, Correlates with the Malignancy of Prostate Cancer. J. Biol. Chem. 2001, 276, 19461–19468.
- Zhuang, L.; bin Peng, J.; Tou, L.; Takanaga, H.; Adam, R.M.; Hediger, M.A.; Freeman, M.R. Calcium-Selective Ion Channel, CaT1, Is Apically Localized in Gastrointestinal Tract Epithelia and Is Aberrantly Expressed in Human Malignancies. Lab. Investig. 2002, 82, 1755–1764.
- Fixemer, T.; Wissenbach, U.; Flockerzi, V.; Bonkhoff, H. Expression of the Ca2+-Selective Cation Channel TRPV6 in Human Prostate Cancer: A Novel Prognostic Marker for Tumor Progression. Oncogene 2003, 22, 7858–7861.
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 Channel Controls Prostate Cancer Cell Proliferation via Ca(2+)/NFAT-Dependent Pathways. Oncogene 2007, 26, 7380–7385.
- Raphaël, M.; Lehen’kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; vanden Abeele, F.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 Calcium Channel Translocates to the Plasma Membrane via Orai1-Mediated Mechanism and Controls Cancer Cell Survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879.
- Lo, Y.C.; Yang, Y.C.; Wu, I.C.; Kuo, F.C.; Liu, C.M.; Wang, H.W.; Kuo, C.H.; Wu, J.Y.; Wu, D.C. Capsaicin-Induced Cell Death in a Human Gastric Adenocarcinoma Cell Line. World J. Gastroenterol. 2005, 11, 6254–6257.
- Jin, J.; Lin, G.; Huang, H.; Xu, D.; Yu, H.; Ma, X.; Zhu, L.; Ma, D.; Jiang, H. Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating P53. Int. J. Biol. Sci. 2014, 10, 285–295.
- Sarkar, A.; Bhattacharjee, S.; Mandal, D.P. Induction of Apoptosis by Eugenol and Capsaicin in Human Gastric Cancer AGS Cells--Elucidating the Role of P53. Asian Pac. J. Cancer Prev. 2015, 16, 6753–6759.
- Kim, M.Y.; Trudel, L.J.; Wogan, G.N. Apoptosis Induced by Capsaicin and Resveratrol in Colon Carcinoma Cells Requires Nitric Oxide Production and Caspase Activation. Anticancer Res. 2009, 29, 3733–3740.
- Xu, S.; Cheng, X.; Wu, L.; Zheng, J.; Wang, X.; Wu, J.; Yu, H.; Bao, J.; Zhang, L. Capsaicin Induces Mitochondrial Dysfunction and Apoptosis in Anaplastic Thyroid Carcinoma Cells via TRPV1-Mediated Mitochondrial Calcium Overload. Cell Signal. 2020, 75, 109733.
- Pawar, J.S.; Mustafa, S.; Ghosh, I. Chrysin and Capsaicin Induces Premature Senescence and Apoptosis via Mitochondrial Dysfunction and P53 Elevation in Cervical Cancer Cells. Saudi J. Biol. Sci. 2022, 29, 3838–3847.
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of Mitochondrial Electron Transport Chain Complexes in Capsaicin Mediated Oxidative Stress Leading to Apoptosis in Pancreatic Cancer Cells. PLoS ONE 2011, 6, e20151.
- Yagi, T. Inhibition by Capsaicin of NADH-Quinone Oxidoreductases Is Correlated with the Presence of Energy-Coupling Site 1 in Various Organisms. Arch. Biochem. Biophys. 1990, 281, 305–311.
- Kishi, T.; Morré, D.M.; Morré, D.J. The Plasma Membrane NADH Oxidase of HeLa Cells Has Hydroquinone Oxidase Activity. Biochim. Biophys. Acta (BBA)—Bioenerg. 1999, 1412, 66–77.
- Kanu, N.; Zhang, T.; Burrell, R.A.; Chakraborty, A.; Cronshaw, J.; Dacosta, C.; Grönroos, E.; Pemberton, H.N.; Anderton, E.; Gonzalez, L.; et al. RAD18, WRNIP1 and ATMIN Promote ATM Signalling in Response to Replication Stress. Oncogene 2016, 35, 4009–4019.
- Ito, K.; Nakazato, T.; Yamato, K.; Miyakawa, Y.; Yamada, T.; Hozumi, N.; Segawa, K.; Ikeda, Y.; Kizaki, M. Induction of Apoptosis in Leukemic Cells by Homovanillic Acid Derivative, Capsaicin, through Oxidative Stress: Implication of Phosphorylation of P53 at Ser-15 Residue by Reactive Oxygen Species. Cancer Res. 2004, 64, 1071–1078.
- Amantini, C.; Ballarini, P.; Caprodossi, S.; Nabissi, M.; Morelli, M.B.; Lucciarini, R.; Cardarelli, M.A.; Mammana, G.; Santoni, G. Triggering of Transient Receptor Potential Vanilloid Type 1 (TRPV1) by Capsaicin Induces Fas/CD95-Mediated Apoptosis of Urothelial Cancer Cells in an ATM-Dependent Manner. Carcinogenesis 2009, 30, 1320–1329.
- Kim, S.; Kang, C.; Chan, Y.S.; Sun, W.H.; Young, D.Y.; Won, S.S.; Park, M.Y.; Kim, E.; Kim, M.; Kim, B.M.; et al. TRPV1 Recapitulates Native Capsaicin Receptor in Sensory Neurons in Association with Fas-Associated Factor 1. J. Neurosci. 2006, 26, 2403–2412.
- Menges, C.W.; Altomare, D.A.; Testa, J.R. FAS-Associated Factor 1 (FAF1): Diverse Functions and Implications for Oncogenesis NIH Public Access. Cell Cycle 2009, 8, 2528–2534.
- Santoni, G.; Caprodossi, S.; Farfariello, V.; Liberati, S.; Amantini, C. Role of Death Receptors Belonging to the TNF Family in Capsaicin-Induced Apoptosis of Tumor Cells. In Role of Capsaicin in Oxidative Stress and Cancer; Springer: Dordrecht, The Netherlands, 2013; pp. 19–46.
- Huh, H.C.; Lee, S.Y.; Lee, S.K.; Park, N.H.; Han, I.S. Capsaicin Induces Apoptosis of Cisplatin-Resistant Stomach Cancer Cells by Causing Degradation of Cisplatin-Inducible Aurora-A Protein. Nutr. Cancer 2011, 63, 1095–1103.
- Meral, O.; Alpay, M.; Kismali, G.; Kosova, F.; Cakir, D.U.; Pekcan, M.; Yigit, S.; Sel, T. Capsaicin Inhibits Cell Proliferation by Cytochrome c Release in Gastric Cancer Cells. Tumour Biol. 2014, 35, 6485–6492.
- Kim, J.M.; Kim, J.D.; Yu, R.; Kim, B.S.; Shin, M.K.; Han, I.S. Effects of Capsaicin on Induction of C-Jun Proto-Oncogene Expression in Fisher-344 Rats by N-Methyl-N’-Nitro-N-Nitrosoguanidine. Cancer Lett. 1999, 142, 155–160.
- Lu, H.F.; Chen, Y.L.; Yang, J.S.; Yang, Y.Y.; Liu, J.Y.; Hsu, S.C.; Lai, K.C.; Chung, J.G. Antitumor Activity of Capsaicin on Human Colon Cancer Cells in Vitro and Colo 205 Tumor Xenografts in Vivo. J. Agric. Food Chem. 2010, 58, 12999–13005.
- Bai, H.; Li, H.; Zhang, W.; Matkowskyj, K.A.; Liao, J.; Srivastava, S.K.; Yang, G.Y. Inhibition of Chronic Pancreatitis and Pancreatic Intraepithelial Neoplasia (PanIN) by Capsaicin in LSL-KrasG12D/Pdx1-Cre Mice. Carcinogenesis 2011, 32, 1689–1696.
- Ghosh, A.K.; Basu, S. Tumor Macrophages as a Target for Capsaicin Mediated Immunotherapy. Cancer Lett. 2012, 324, 91–97.
- Islam, A.; Hsieh, P.-F.; Liu, P.-F.; Chou, J.-C.; Liao, J.-W.; Hsieh, M.-K.; Chueh, P.J. Capsaicin Exerts Therapeutic Effects by Targeting TNOX-SIRT1 Axis and Augmenting ROS-Dependent Autophagy in Melanoma Cancer Cells. Am. J. Cancer Res. 2021, 11, 4199.
- Zhang, R.; Humphreys, I.; Sahu, R.P.; Shi, Y.; Srivastava, S.K. In Vitro and in Vivo Induction of Apoptosis by Capsaicin in Pancreatic Cancer Cells Is Mediated through ROS Generation and Mitochondrial Death Pathway. Apoptosis 2008, 13, 1465–1478.
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Devaki, T. Capsaicin Provokes Apoptosis and Restricts Benzo(a)Pyrene Induced Lung Tumorigenesis in Swiss Albino Mice. Int. Immunopharmacol. 2013, 17, 254–259.
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Devaki, T. Lysosomal Abnormalities during Benzo(a)Pyrene-Induced Experimental Lung Carcinogenesis—Defensive Role of Capsaicin. Fundam. Clin. Pharmacol. 2009, 23, 97–103.
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Devaki, T. Effect of Capsaicin on Glucose Metabolism Studied in Experimental Lung Carcinogenesis. Nat. Prod. Res. 2009, 23, 763–774.
- Anandakumar, P.; Jagan, S.; Kamaraj, S.; Ramakrishnan, G.; Titto, A.A.; Devaki, T. Beneficial Influence of Capsaicin on Lipid Peroxidation, Membrane-Bound Enzymes and Glycoprotein Profile during Experimental Lung Carcinogenesis. J. Pharm. Pharmacol. 2008, 60, 803–808.
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Asokkumar, S.; Naveenkumar, C.; Raghunandhakumar, S.; Devaki, T. Capsaicin Inhibits Benzo(a)Pyrene-Induced Lung Carcinogenesis in an in Vivo Mouse Model. Inflamm. Res. 2012, 61, 1169–1175.
- Yoshitani, S.I.; Tanaka, T.; Kohno, H.; Takashima, S. Chemoprevention of Azoxymethane-Induced Rat Colon Carcinogenesis by Dietary Capsaicin and Rotenone. Int. J. Oncol. 2001, 19, 929–939.
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2008, 22, 659–661.
- He, J.; Wu, X.; Xie, Y.; Gao, Y.; McClements, D.J.; Zhang, L.; Zou, L.; Liu, W. Capsaicin Encapsulated in W/O/W Double Emulsions Fabricated via Ethanol-Induced Pectin Gelling: Improvement of Bioaccessibility and Reduction of Irritation. Int. J. Biol. Macromol. 2023, 235, 123899.