Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The transcription factor Nrf2 (nuclear factor [erythroid-derived 2]-like 2) is of major importance as the defense instrument against OS and alters anti-inflammatory activities related to different pathological states.
- Nrf2 activation
- autoimmune diseases
- inflammation
- autoimmunity
1. Introduction
Under normal conditions, the immune system protects and guards the body against attacks and infections, which are possible sources of diseases and syndromes. However, if the complex immune system regulatory network breaks down, it misguidedly attacks healthy cells, tissues, and organs. The malfunctioning of the immune system may result in attacks on any part of the body, weakening bodily functions and sometimes turning life-threatening [1]. Oxidative stress (OS) is described as a modification in the redox state [2] and the production of free radicals in cells in response to an environment of inherited traits. The immunological progression in cells may promote OS and, furthermore, the accumulation of OS worsens the pathophysiology of the disorder [3]. The over-creation of both reactive oxygen (ROS) and nitrogen (RNS) species has been associated with numerous autoimmune disorders [4]. Among the enzymes in human cells that are mainly involved in the production/bioavailability of radical species, those that are particularly relevant are NADPH oxidases (NOXs), mitochondrial electron transport chain complexes, nitric oxide synthases (NOSs), xhantine oxidases (XOs), and the hydrogen sulfide-producing enzymes cystathionine-β-synthase and cystathionine-γ-lyase [5]. ROS can actively influence both innate and adaptive immunity by regulating the response of its components at different levels, such as antigen production and apoptotic cell clearance [6]. The chemical and post-translational alterations of proteins that occur in a pro-oxidant environment may occasionally promote the formation of “neoepitopes”, which are recognized as “foreign” by the immune system and trigger the development of autoantibodies, which are made against substances formed by a person’s own body.
The Kelch-like ECH-associated protein 1–nuclear factor (erythroid-derived 2)-like 2 (KEAP1-Nrf2) structure has a significant role in antioxidant reactions, and associates to protect cells from numerous redox instabilities [7][8]. The Nrf2 protein in humans is 605 amino acids long and possesses seven highly conserved areas known as Nrf2-ECH homology (Neh) domains (Figure 1). Neh1 includes the CNC–bZIP domain, which moderates heterodimerization with Maf [9]. The Neh2 domain has two degrons that are, in particular, attached by Keap1.
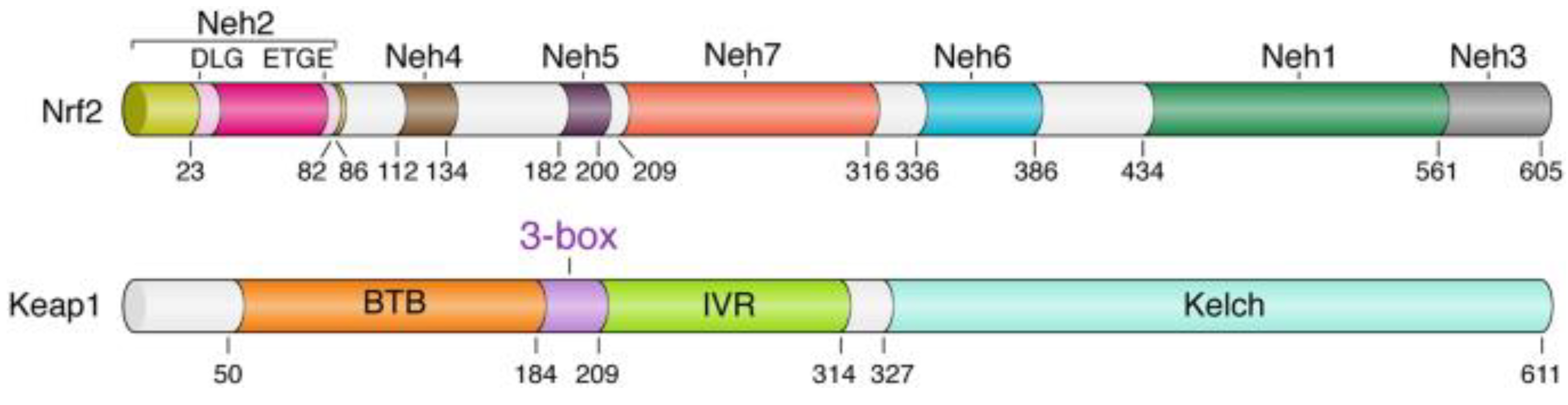
Figure 1. Domain architecture of the Keap1 and Nrf2 proteins. Domain boundaries and residue numbers are shown for human proteins [10].
Normally, Nrf2 is controlled by KEAP1. Once cells are affected by antioxidants, Nrf2 migrates into the nucleus and increases the expression of many of the genes that encode antioxidant proteins and detoxifying enzymes [11]. Nrf2 is known to be a fundamental controller of cellular defense, inflammation, and redox balance renewal [12]. Nrf2 controls the expression of over 200 genes, which are included in the promoter area called the antioxidant response element (ARE). The genes controlled by Nrf2 encode enzymes that contribute to endobiotic/xenobiotic metabolism, OS/inflammatory reactions, carbohydrate/lipid metabolism, and protein degradation [13][14].
In the course of OS, ROS interacts with the cysteine amino acid on Keap1, which modifies the formation of the Keap1–Nrf2 compound and, thus, escapes the degradation of Nrf2 [15]. For this reason, freshly gathered Nrf2 proteins gather in the cytoplasm and are then transferred to the nucleus, where the Nrf2 binds to the ARE. This supports the transcription of numerous target genes, like the detoxifying enzymes heme oxygenase (Hmox1), NAD(P)H dehydrogenase 1 (Nqo1), and sulfiredoxin 1 (Srxn1); the xenobiotic metabolizing enzymes; as well as the genes taking part in glutathione (GSH) production, such as glutathione reductase (GSR) and glutamate–cysteine ligase modifier (GCLM) [16].
The regulation of Nrf2 primarily occurs through the managed maintenance of Nrf2 protein levels. There are three E3 ubiquitin ligase complexes that are responsible for the ubiquitylation and degradation of Nrf2: the CUL3-RBX1-KEAP1 complex, the SCF/β-TrCP complex, and HRD1. Each of them mediates the degradation of Nrf2 in response to various stimuli in specific subcellular compartments. The CUL3-RBX1-KEAP1 complex responds to electrophilic/oxidative stress in the cytosol. The nuclear or cytosolic SCF/β-TrCP complex is more susceptible to metabolic shifts and is regulated by GSK3-β. HRD1 is localized to the ER and has only been shown to ubquitylate Nrf2 during ER stress [17]. It is essential to state that other signaling paths, epigenetic factors, and post-translational modifications also regulate Nrf2. Similarly, the activation or inhibition of the Nrf2 path can be achieved by targeting the negative regulation of Nrf2 (Figure 2).
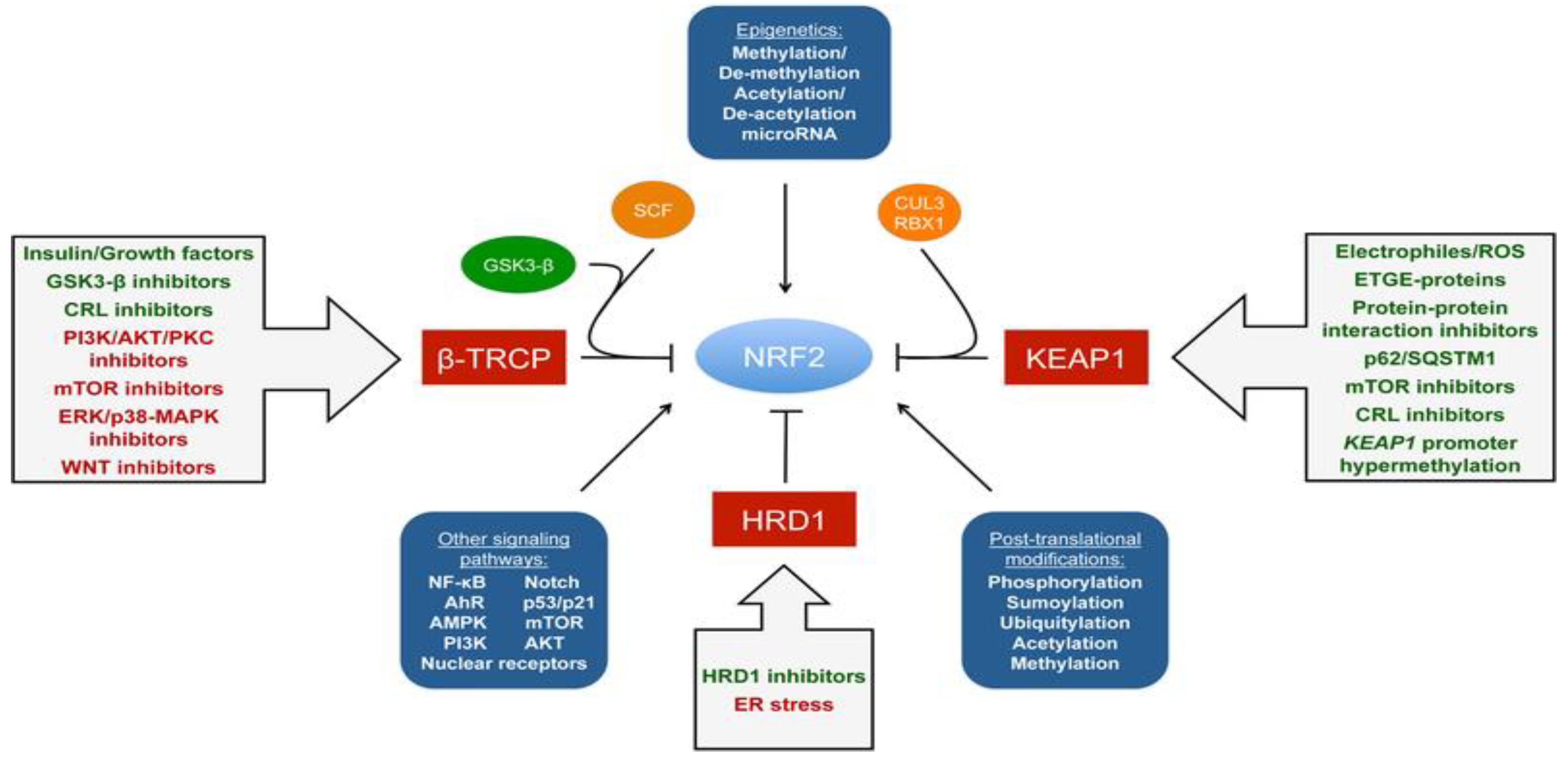
Figure 2. Regulation of NRF2 and possible modes of activation [18].
The molecular activation and cytoprotective activity of the KEAP1-NRF2 route comprise four separate but interlinked elements:
- (a)
-
The chemical inducers of NRF2 activity (e.g., tBHQ, CDDO-Im, and TBE-31 [19]) (Table 1);Table 1. Inducers of Nrf2 activity [20].
Inducer Example Endogenous signaling compounds/metabolites H2O2
Lipid peroxidation products
Nitric oxide
8-Nitro-cGMP
Hydrogen sulfide
MethylglyoxalOncometabolites Fumarate
SuccinylacetoneImmunometabolite Itaconate Dietary compounds Sulforaphane
CurcuminPharmaceuticals Dimethyl fumarate
BardoxyloneMicroorganisms Bacteria/lipopolysaccharide
Marburg virus
Plasmodium infectionExtracellular inducers Heat
Laminar flow
UVA radiation
Exercise - (b)
-
KEAP1, the protein sensor of these inducers;
- (c)
-
The transcription factor NRF2, which regulates the transcriptional response to inducers and oxidative stress;
- (d)
-
The target genes that supply the cytoprotective output of the path [20].
There is a developing theme in the Nrf2 area that targeting Nrf2 in disorder is both context and time-dependent. Harnessing the advantageous outcomes of pharmacological activation of Nrf2 is an essential part of Nrf2-based chemoprevention and intervention in other chronic illnesses, such as neurodegeneration, diabetes, cardiovascular disease, autoimmune diseases, and chronic kidney and liver disease. Nevertheless, a growing number of investigations have indicated that Nrf2 is already elevated in specific cancer and disease steps, suggesting that pharmacological agents developed to mitigate the potentially destructive or transformative results associated with protracted activation of Nrf2 should also be considered. Instances of current Nrf2 activators and inhibitors, as well as Nrf2 expression grades in disorders, are outlined in Figure 3 [18]. Nrf2-targeted therapeutics are shown in Table 2.
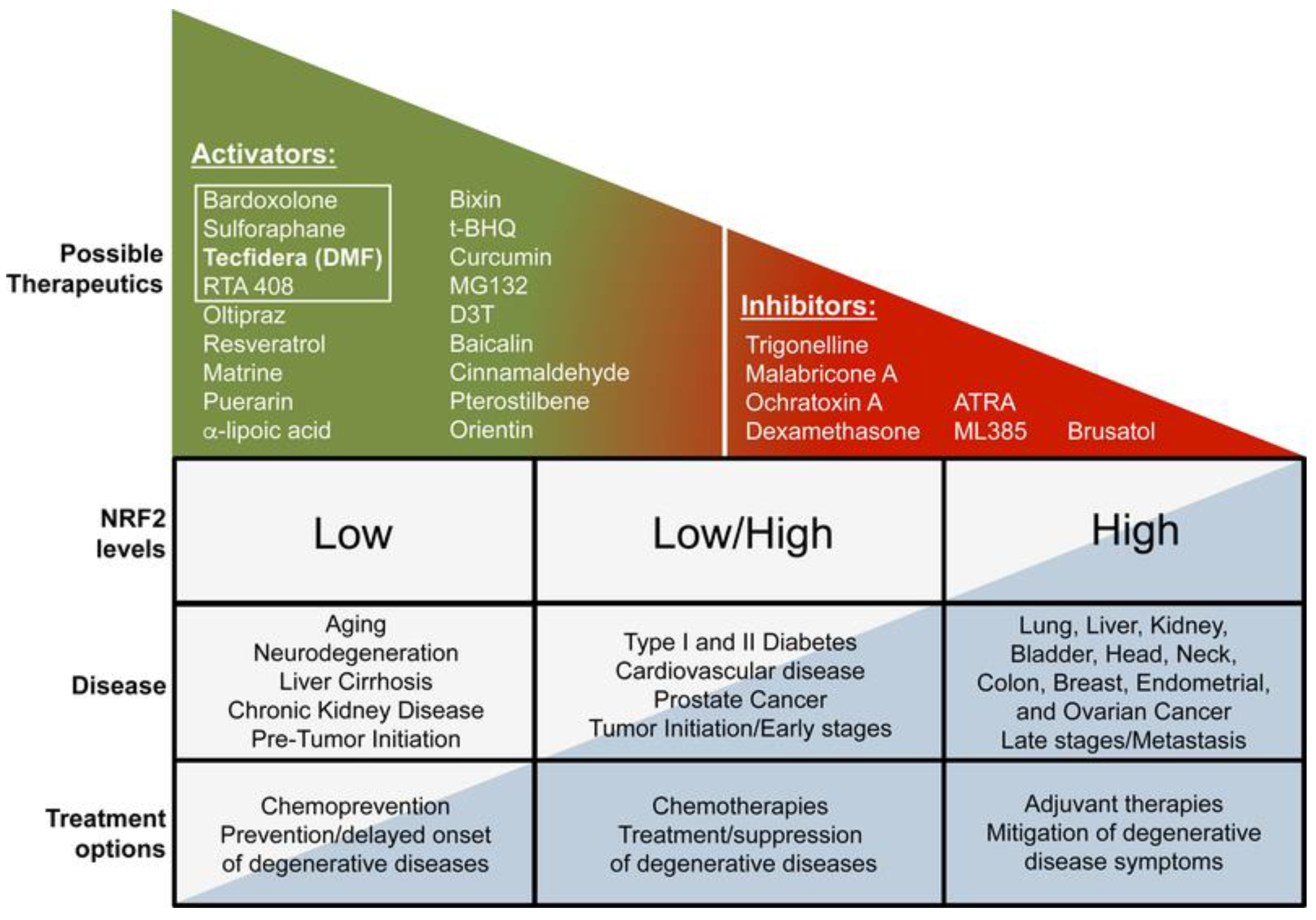
Figure 3. Nrf2-targeted therapeutics [18].
Table 2. Nrf2-targeted therapeutics [21].
| Therapeutics | Mechanism of Action | Molecules | |
|---|---|---|---|
| Electrophilic NRF2 activators | Natural products | Electrophilic modification of KEAP1-C151 | Sulphorapane Bixin |
| Natural product-derived | Electrophilic modification of KEAP1-C151 | Dimethyl fumarate Bardoxolone methyl |
|
| Pro-electrophilic NRF2 activators | Natural products | Electrophilic modification of KEAP1-C151 | Carnosic acid Carnosol |
| Non-electrophilic compounds | Peptides | Binding to KEAP1 Kelch domain | Ac-DPETGEL-OH (7mer) FITC-β-DEETGEF-OH (7mer) FITC-LDEETGEFL-NH2 (9mer) FAM-LDEETGELP-OH (9mer) |
| Small molecules | Binding to KEAP1 Kelch domain | Compound 2 Cpd15 Cpd16 (SRS)-5 AN-465/144580 |
|
| KEAP1-independent NRF2 activators | Natural products | GSK-3 inhibition | Nordihydroguaiaretic acid |
| Synthetic | HRD1 inhibition | LS-102 | |
| NRF2 inhibitors | Natural products | Global protein translation inhibitor | Brusatol |
Ac: acetyl, FITC: fluoresceine isothiocyanate, FAM: carboxyfluoresceine.
The cellular protection ability supported by Nrf2 activation seems to maintain the stability of CD4+ and CD8+ of lymph node cells for appropriate essential immune reactions. Nrf2 is able to able to negatively regulate pro-inflammatory signaling molecules like p38 MAPK, NF-kB, and AP-1. Another task of Nrf2 is to prevent the formation of pro-inflammatory intermediates such as cytokines, chemokines, cell adhesion molecules, COX-2, matrix metalloproteinases, and iNOS. Even though not openly clarified, the antioxidant activity of genes directed by Nrf2 might supportively control the innate immune defense [22].
It seems that Nrf2 action is crucial in regulating cellular mechanisms regarding the determination of inflammatory progression. To accomplish this, Nrf2 interacts with nuclear factor-κB (NF-κB) via various chemical connections [23]. The phosphorylation reaction of NF-κB inhibitor (IκBα) by IκB kinase (IKKβ) leads to IκBα deprivation, which causes nuclear translocation and DNA attachment to NF-κB. Nrf2 reduces NF-κB stimulation by reacting with Keap1. Nrf2 is displayed to extensively regulate the immune defense by acting with vital innate immune elements such as the toll-like receptors–Nuclear factor kappa B (NF-κB) cascade, inflammasome signaling, and the type-I interferon reaction.
The role of Nrf2 in downgrading inflammation has been recognized in some animal studies with different pathological disorders [24]. Furthermore, Nrf2 absence worsens autoimmune disorders like rheumatoid arthritis (RA) [25] and systemic lupus erythematosus (SLE) [26], whereas Nrf2 activation improves autoimmune encephalomyelitis [27].
Growing indications have reinforced the idea that the regulatory role of Nrf2 is not limited to OS but is also directed to inflammation mechanisms, immune system disorders, and cartilage and bone metabolism [28]. Research has shown that Nrf2 activation might regulate diverse progressions and intermediates that take place in the pathology of autoimmune disorders such as RA, systemic SLE, osteoarthritis (OA), and osteoporosis. Nrf2 activation, through multiple pathways, induces a strong antioxidant response associated with beneficial effects against various conditions (Figure 4). The goal of this observation is to emphasize Nrf2 as a novel pharmacological target in above mentioned illnesses.
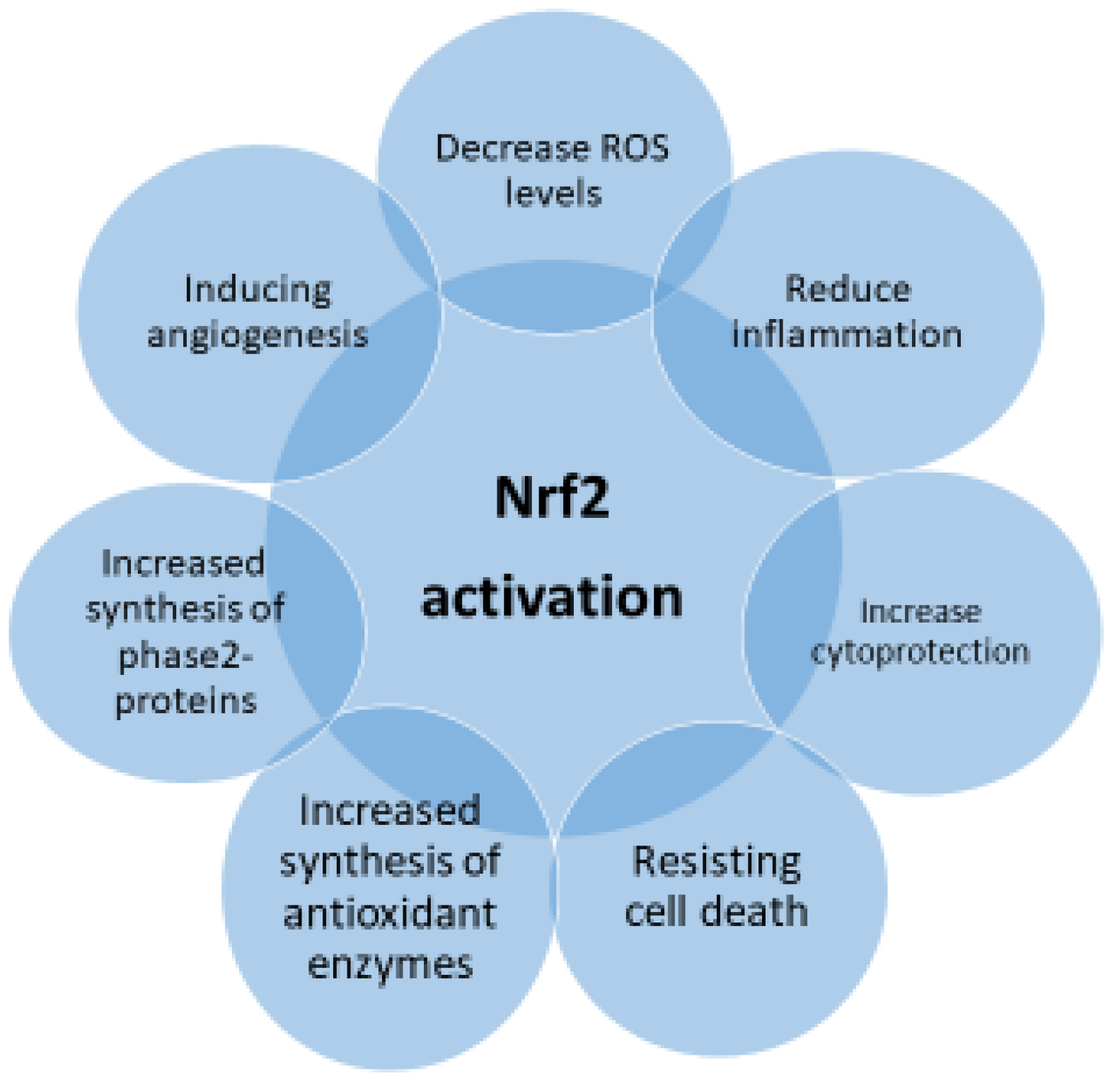
Figure 4. Pharmacological properties of Nrf2 activation.
2. Nrf2 Regulation and Inflammation
Given the extensive display of stimuli that trigger Nrf2 and the various cellular operations that it handles, the regulation of Nrf2 activity is complicated and multifactorial. Absolutely, Nrf2 activation can be managed at the transcriptional and post-transcriptional level via the regulation of protein stability, post-transcriptional shifts, and the availability of binding fellows (Figure 5).
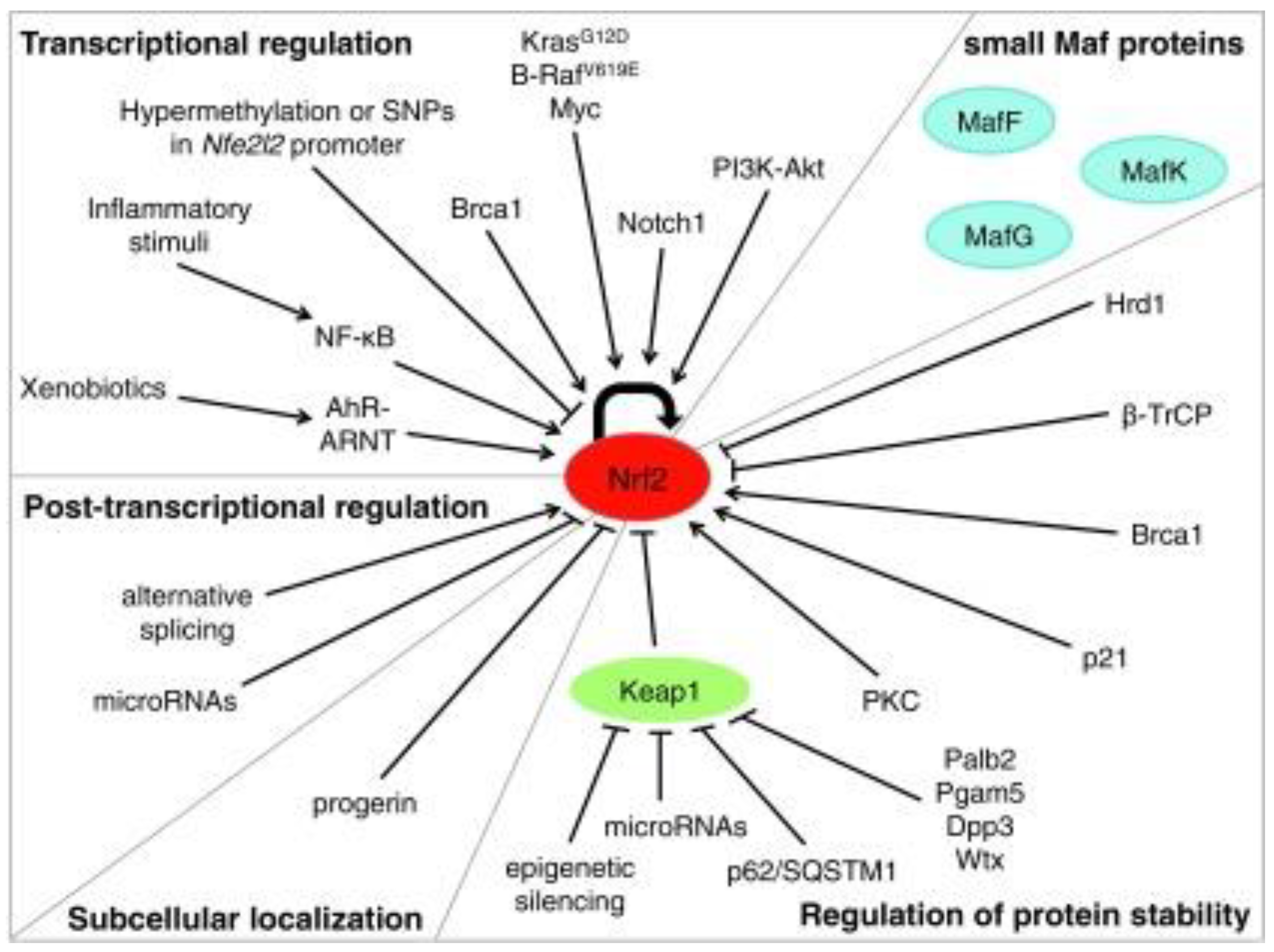
Figure 5. Mechanisms of regulation of Nrf2 activity [29].
Inflammation is the most known consequence of numerous immune disorders and illnesses. Research has shown that Nrf2 is linked to anti-inflammatory progression by altering the employment of inflammatory cells and by modifying gene expression at the ARE level. The Nrf2 protein half-life is less than 20 min in the cell [30]. The Keap1/Nrf2/ARE signaling pathway primarily controls anti-inflammatory gene expression and prevents the development of inflammation in many diseases [31]. Other signaling pathways like NF-κB, MAPK (mitogen-activated protein kinase), and JAK (janus kinase)-STAT (signal transducers and activators of transcription) are considered to be of relevance in the progress of inflammation [32]. It was discovered that Nrf2 controls the phase II detoxifying enzymes involving NADPH, NAD(P)H quinone oxidoreductase 1, glutathione peroxidase, ferritin, heme oxygenase-1 (HO-1), and antioxidant genes that protect tissues against damage with their anti-inflammatory properties [33].
The main role of inflammation is to eliminate the foundation of disorder and re-establish homeostasis. Conventionally, native immunity is the prompt reaction in the procedure of phagocytosis, while adaptive immunity is antigen-reliant and described by an immunological recall that allows for increasing an effective immune response following contact with a similar antigen. The symbol of the inflammatory process is the formation of signaling elements named cytokines [34]. Activation of Nrf2 in several diseases related to OS and inflammation lessens markers of damage and inhibits illness development. These properties are thought to be the result of the upregulation of antioxidative and phase II detoxifying enzymes by Nrf2 as well as the straight role of control in the formation of inflammation. It is known that Nrf2 prevents transcription of pro-inflammatory IL-6 and IL-1β cytokines by making bonds to genes in macrophages and stopping RNA Pol II employment [12].
Because Nrf2 is a lead regulator of redox homeostasis, it wields indirect control on NF-kB activity. Lipopolysaccharides (LPSs) trigger simultaneously a fast, pro-inflammatory NF-kB response and a slow Nrf2 reaction. The NF-kB response is subsequently inhibited while Nrf2 is maximally functional [35]. For example, Ras-related C3 botulinum toxin substrate 1, a small G protein of the Rho family, activated the NF-kB path and Nrf2 overexpression obstructed, whereas Nrf2 knockdown improved NF-kB-dependent transcription [35]. Invariably, in Nrf2-deficient (Nrf22/2) mice challenged with LPS or tumor necrosis factor (TNF)-a, the activity of IKK was aggravated and led to raised phosphorylation and degradation of IkB [36]. Nrf2 furthermore generates an anti-inflammatory phenotype that modulates the functions of CD8+ T cells [37] as well as in macrophages and microglia [38][39][40]. This is because Nrf2 augments cysteine and GSH levels in macrophages via regulation of the cystine/glutamate transporter and the GSH-synthesizing enzyme g-glutamyl cysteine ligase modulator and catalytic subunits (g-glutamyl cysteine ligase modulator subunit (GCLM) and g-glutamyl cysteine ligase catalytic subunit (GCLC)). Contrarily, a lack of GSH sensitizes macrophages to Nrf2 activation by LPS [41]. All these investigations indicate Nrf2 as an anti-inflammatory factor critical in maintaining the intensity and period of inflammatory reactions (Figure 6).
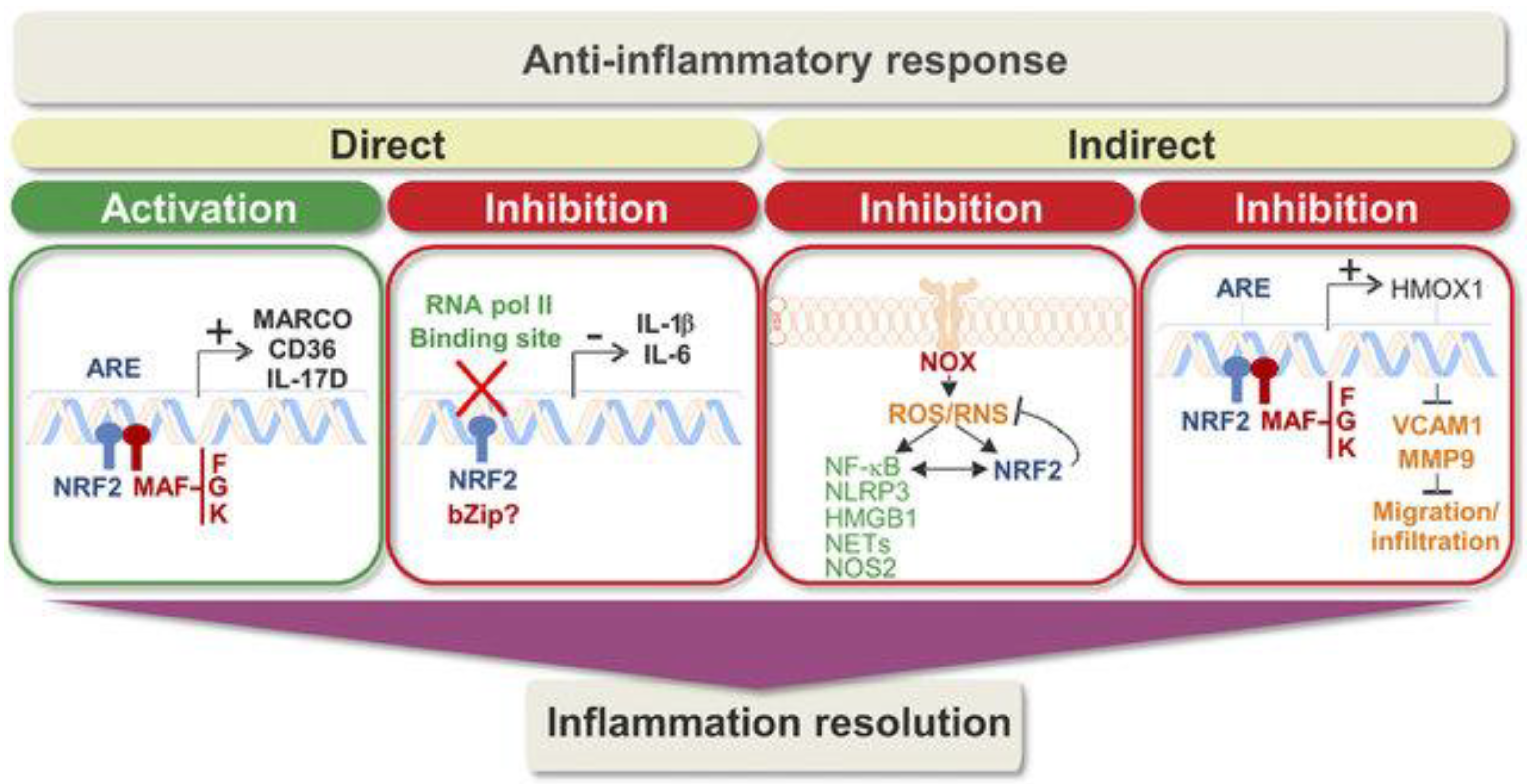
Figure 6. Direct and indirect regulation of inflammation by Nrf2 [40].
Direct mechanisms of activity contain transcriptional induction of anti-inflammatory genes as well as transcriptional repression of pro-inflammatory genes. In the second box, the question mark reveals that additional work is needed to determine the bZip partner of Nrf2 in this function, if any. Indirect mechanisms to compensate for inflammation include ROS/RNS modulation and inhibition of the migration/infiltration of immune cells. In general, these paths guide an anti-inflammatory reaction that properly resolves inflammation. The presence of polymorphisms in NFE2L2 related to diminished transcriptional activity, the varied levels of target genes in patients, and promising data from preclinical analyses support a suitable position of Nrf2 in inflammation resolution [42].
Carbon monoxide (CO) is a steady gaseous molecule that reacts selectively with transition metals in a distinct redox state, and these characteristics limit the interaction of CO with described biological targets that transduce its signaling action [43]. Because of the increased affinity of CO for ferrous heme, these targets can be categorized into heme-containing proteins, illustrating an enormous assortment of sensors and enzymes with a sequence of various roles in cells and organisms. Despite this concept, advancement in specifying which of these targets are selective for CO has been slow, and even the significance of raised carbonmonoxy hemoglobin, a classical marker utilized to diagnose CO poisoning, is not well-comprehended. Nonetheless, the usage of CO gas and CO-releasing molecules as pharmacological strategies in models of disease has supplied new vital knowledge about the signaling properties of CO. CO is continually yielded by heme oxygenases in mammalian cells during heme degradation [44].
Heme oxygenases exist in constitutive (HO-2) and inducible (HO-1) isoforms and are derivatives of two different genes, HMOX2 and HMOX1. While heme oxygenase-2 (HO-2) has separate tissue localization, being predominantly expressed in the testes, brain, and endothelium, heme oxygenase-1 (HO-1) is upregulated in all tissues investigated, pursuing several types of stress stimuli involving oxidative stress, which is an underlying factor in various pathological conditions [44].
HO-1, also known as heat shock protein 32, is the rate-limiting and inducible cytoprotective enzyme in the heme degradation path that degrades heme into free iron, carbon monoxide, and biliverdin, which is then rapidly converted into bilirubin [45][46]. Biliverdin is henceforward reduced to bilirubin via biliverdin reductase. Both biliverdin and bilirubin are bile pigments with antioxidant effects [47]. Endogenous CO can function as a second messenger, thereby affecting a variety of physiological and pathological processes involving cell proliferation, inflammation, apoptosis, and injury [48]. As a cytoprotective enzyme, HO-1 serves an essential function in controlling cell homeostasis.
Under miscellaneous pathophysiological stress or stimulation conditions, such as hypoxia, ultraviolet light, inflammatory mediators, heme, ischemia, and other harmful stimuli, HO-1 expression is generated to guard cells against oxidative and inflammatory harm [49]. In the presence of the forenamed stimulatory elements, the yielded expression of HO-1 is mainly influenced by redox-sensitive transcription factors, involving Nrf2, activator protein 1 (AP1), hypoxia-inducible factor (HIF), and BTB and CNC homology 1 (Bach1) [50].
The association between Nrf2 and the induction of HO-1 is well established and is conditional on the existence of antioxidant reaction elements in the promoter of the HMOX1 gene [51][52].
Nrf2 is now known as the lead regulator of cellular antioxidant protection systems because it handles, in addition to HO-1, the expression of a battery of detoxification enzymes, such as NAD(P)H dehydrogenase quinone 1, glutathione S-transferases, and peroxiredoxins [53][54][55]. Therefore, the defense wielded by Nrf2 is reliant on these genes, and their silencing, to a significant capacity, switches the helpful actions of Nrf2 activation [56]. It is not surprising then, that many of the HO-1 inducers that have been represented by various authors over the last decade seem to include Nrf2 as the upstream factor inducing this response [57].
It is also fascinating to report that the transcription factor Nrf2 was significantly augmented for the genes positively associated with HMOX1. The Nrf2-Keap1-HMOX1 path is a cellular defense mechanism playing a vital role in shielding against oxidative stress and inflammation [58]. This pathway is activated in response to diverse stimuli, involving reactive oxygen species (ROS), heavy metals, and xenobiotics. Once triggered, it leads to the transcriptional upregulation of antioxidant and detoxifying genes involving HMOX1 [59][60].
Mangano et al. [61] designed a study to explore the immunoregulatory mechanisms operating in the development and regulation of concanavalin A (ConA)-induced hepatitis. They evaluated the role of the anti-inflammatory path Nrf2/HO-1/CO in this condition and investigated the in vivo administration of CO through the CO-releasing molecule (CORM). They observed that the Nrf2/HO-1/CO pathway is fundamental for immune response regulation. Also, Nikolic et al. [62] tried to find efficacy and the mechanisms of action of the CO-releasing molecule (CORM)-A1 in preclinical models of type 1 diabetes. Their data indicated that the capability of CORM-A1 to save mice from developing type 1 diabetes supplies useful evidence of conception for the probable exploitation of controlled CO delivery in clinical settings for the therapy of autoimmune diabetes.
In the absence of Nrf2, oxidative cell injury and apoptosis may increase the formation of autoantigens, leading to the triggering of T cells and the creation of autoantibodies by B cells. Furthermore, the lack of phase II enzymes results in an increase in ROS. Nrf2 is a chief controller of cellular defense reactions to oxidation, and it is expected that Nrf2 activation is able to defend against OS associated with autoimmune pathogenesis [27][28].
Studies propose that Nrf2 responds to the NF-κB-associated inflammation reaction by challenging the transcription co-activator cAMP response element (CREB) binding protein (CBP) [63]. The histone acetylation reaction and subsequent DNA transcription are controlled by CBP-p300. CBP was stated to interrelate with the domains Neh4 and Neh5 of Nrf2, causing the acetylation of Neh1, which is directly involved in DNA binding [64]. It was described that the link between the N-terminal area of the p65 subunit of NF-κB and Keap1 could stop the Nrf-2 cascade. Nevertheless, various studies reported that diverse causes of OS usually stimulate both NF-κB and Nrf2-ARE signaling [65][66].
Inflammation is a multifaceted interaction among several inflammatory cells, which gives many signaling agents like arachidonic acid type compounds, phospholipid mediators, and cytokines that seem to have an essential part in some inflammatory responses, influencing the reactions between pro- and anti-inflammatory systems that lead to various disorders [67]. The activation of the Nrf2 pathway determines a remarkable event to address and direct the evolution of inflammation. It was described that the activation of Nrf2 inhibits LPS-induced modulation of pro-inflammatory cytokines including IL-6 and IL-1β. This relationship was confirmed by the finding that Nrf2 controls NF-κB-oriented transcription of pro-inflammatory cytokine genes [11].
Mills et al. [68] described the effects of a novel itaconate derivative, 4-octyl itaconate, that protects mortality in vivo and reduces cytokine formation. They showed that type I interferons increase the expression of Irg1 (also known as Acod1) and itaconate formation. In addition, they found that this reaction confines the type I interferon response. Their data show that itaconate is an important anti-inflammatory molecule that limits inflammation through Nrf2 and modulates type I interferons.
Yan et al. [69] found that treatment with dimethyl fumarate considerably enhanced cognitive insufficiencies and partly inverted neuronal injury in the hippocampus triggered by chronic cerebral hypoperfusion (CCH). In addition, this management reduced the concentration of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-6 in the hippocampus and mediated NF-κB signaling. The results suggest that dimethyl fumarate may increase cognitive injury in rats with CCH, undoubtedly by lessening inflammation and ferroptosis of neurons.
Ding et al. [70] reported that Nrf2 absence considerably raised IL6 and IL10 secretion by M1 macrophages. The control of these macrophage inflammations via Nrf2 shows numerous roles for Nrf2 in modifying inflammation in macrophages. A lack of Nrf2 augmented the Glu4 protein level and reduced AKT and GSK3β protein phosphorylation in M1 macrophages, signifying multiple roles for Nrf2 in modifying glucose metabolism in macrophages. These results back the perspective that Nrf2 is a pharmacological target for the inhibition and cure of inflammation and obesity-linked disorders such as diabetes and atherosclerosis.
Numerous in vivo and in vitro experiments have revealed the result of Nrf2 in retreating diabetes mellitus by responding to the progression of OS [71]. Nrf2 expression has been found to be induced under OS conditions. Therefore, there is an urgent need for research and clinical trials to develop essential therapeutics to protect against the progression of diabetes and to upregulate genes contained in Nrf2 as a means of combating hyperglycemia [72]. In addition, Nrf2 functions as a chief factor in detoxifying cellular reactions that deliver sufficient defense against OS and damage. Several lines of evidence point to the vital act of OS in diabetes. A similar theory also shows that an increase in ROS significantly causes the growing link between free fatty acids and hyperglycemia. Furthermore, ROS activates stress-sensitive signaling pathways, ultimately leading to diabetes mellitus, β-cell dysfunction, and insulin resistance [73][74].
As mentioned above, the upregulation of specific Nrf2 target proteins such as glutathione S-transferase, glutamyl cysteine synthase, quinone oxidoreductase, and heme oxygenase-1, takes place through specific elements that represent an antioxidant reaction and are located in the promoters of these genes. This synchronized act of modified genes encoding antioxidant, anti-inflammatory, and detoxifying regulators functions as possible healer compounds to protect against the increased OS and inflammation in diabetes mellitus [75].
Ferroptosis is a type of cell death mechanism that takes place intracellularly in the presence of iron. This system is a different action from cell apoptosis, necrosis, and autophagy, and it is defined by an imbalanced redox system and augmented amounts of intracellular ROS [76]. Li et al. [77] showed that ferroptosis is involved in the development of diabetic nephropathy, which is possibly a consequence of reactions between metabolic and hemodynamic mechanisms. It seems that the metabolic and hemodynamic disorders found in diabetes interrelate with ROS formation. Gene regulation and stimulation of transcription factors are affected by contacts between metabolic inducements, hemodynamic issues, and several ROS in diabetes [78]. The upregulation of Nrf2 by fenofibrate treatment inhibited diabetes-related ferroptosis and delayed the progression of diabetic nephropathy. Li and his colleagues’ research revealed the mechanism of the development of diabetic nephropathy from a new perspective and provided a new approach to delaying the progression of diabetic nephropathy.
There are several reports on the positive effects of sulforaphane, extracted from broccoli sprouts, on macrovascular complications in diabetes [79][80][81][82][83]. Dh404 is a bardoxolone methyl derivative that has been used clinically for the management of diabetic nephropathy. Dh404 triggers Nrf2 by an alteration of KEAP1, a reaction similar to sulforaphane [84]. Tan et al. [85] reported that Dh404 reduced atherosclerosis in diabetic conditions at lower doses with a reduction in OS and inflammatory factors. Dimethyl fumarate is a recognized Nrf2 activator and is used clinically for the management of multiple sclerosis. The study by Ha et al. [86] suggests a possible defensive effect of dimethyl fumarate on macrovascular complications in diabetes. Tert-butyl hydroquinone stimulates Nrf2 by directing Cys-151 in the KEAP1 protein [19]. It was shown to improve diabetes-related atherosclerosis in an animal study [87]. It was found to increase Nrf2 action in macrophages in atherosclerotic lesions and promote autophagic activity. This resulted in a reduction in atheroma plaque size, expansion, and lipid content, as well as in decreased lesional macrophages, foam cell size, and chemokine expression.
Yu et al. [88] suggested that high uric acid levels trigger ferroptosis of macrophages in the development of atherosclerosis. Additionally, promoting autophagy and preventing ferroptosis by triggering Nrf2 might ameliorate atherosclerosis caused by elevated uric acid concentrations. These results may provide a better understanding of the role of high uric acid concentrations in the development of atherosclerotic plaques and show a pharmacological approach for atherosclerotic vascular disease connected with high uric acid levels.
In contrast, Li et al. [89] indicated that Nrf2 deficiency is associated with a reduction in atherosclerotic plaques and may reduce physiopathological development by attenuating lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1)-mediated production and relocation of vascular smooth muscle cells.
Zhao et al. [90] show that melatonin may be effective in protecting against smoking-related vascular damage and atherosclerosis through the Nrf2/ ROS /NLRP3 cascade. Generally, these findings deliver convincing support for the use of melatonin clinically to reduce inflammatory vascular damage and atherosclerosis caused by smoking.
Feng et al. [91] demonstrated the positive characteristics of kaempferol against postmenopausal atherosclerosis related to the PI3K/AKT/Nrf2 pathway facilitated by G protein-coupled oestrogen receptor (GPER) activation.
This entry is adapted from the peer-reviewed paper 10.3390/brainsci13111532
References
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301.
- Gong, D.-J.; Wang, L.; Yang, Y.-Y.; Zhang, J.-J.; Liu, X.-H. Diabetes Aggravates Renal Ischemia and Reperfusion Injury in Rats by Exacerbating Oxidative Stress, Inflammation, and Apoptosis. Ren. Fail. 2019, 41, 750–761.
- Ramani, S.; Pathak, A.; Dalal, V.; Paul, A.; Biswas, S. Oxidative Stress in Autoimmune Diseases: An Under Dealt Malice. Curr. Protein Pept. Sci. 2020, 21, 611–621.
- Mehling, R.; Schwenck, J.; Lemberg, C.; Trautwein, C.; Zizmare, L.; Kramer, D.; Müller, A.; Fehrenbacher, B.; Gonzalez-Menendez, I.; Quintanilla-Martinez, L.; et al. Immunomodulatory role of reactive oxygen species and nitrogen species during T cell-driven neutrophil-enriched acute and chronic cutaneous delayed-type hypersensitivity reactions. Theranostics 2021, 11, 470–490.
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14.
- Lin, W.; Shen, P.; Song, Y.; Huang, Y.; Tu, S. Reactive Oxygen Species in Autoimmune Cells: Function, Differentiation, and Metabolism. Front. Immunol. 2021, 25, 635021.
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140.
- Telkoparan-Akillilar, P.; Panieri, E.; Cevik, D.; Suzen, S.; Saso, L. Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules 2021, 26, 1417.
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1–Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384.
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107.
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624.
- Barati, M.T.; Caster, D.J. The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus. Metabolites 2022, 12, 151.
- Hayes, J.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218.
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Activation of Nrf2 by Natural Bioactive Compounds: A Promising Approach for Stroke? Int. J. Mol. Sci. 2020, 21, 4875.
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717.
- Hiemstra, S.; Fehling-Kaschek, M.; Kuijper, I.A.; Bischoff, L.J.M.; Wijaya, L.S.; Rosenblatt, M.; Esselink, J.; van Egmond, A.; Mos, J.; Beltman, J.B.; et al. Dynamic modeling of Nrf2 pathway activation in liver cells after toxicant exposure. Sci. Rep. 2022, 12, 7336.
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; Rojo de la Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686.
- Dodson, M.; Rojo de la Vega, M.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in disease: Timing is everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575.
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146.
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20.
- Rojo de la Vega, M.; Dodson, M.; Chapman, E.; Zhang, D.D. NRF2-targeted therapeutics: New targets and modes of NRF2 regulation. Curr. Opin. Toxicol. 2016, 1, 62–70.
- Kim, J.; Surh, Y.J. The Role of Nrf2 in Cellular Innate Immune Response to Inflammatory Injury. Toxicol. Res. 2009, 25, 159–173.
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626.
- Suzen, S.; Tucci, P.; Profumo, E.; Buttari, B.; Saso, L. A Pivotal Role of Nrf2 in Neurodegenerative Disorders: A New Way for Therapeutic Strategies. Pharmaceuticals 2022, 15, 692.
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507.
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13.
- Suzuki, T.; Murakami, S.; Biswal, S.S.; Sakaguchi, S.; Harigae, H.; Yamamoto, M.; Motohashi, H. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol. Cell. Biol. 2017, 37, e00063-17.
- Ferrándiz, M.L.; Nacher-Juan, J.; Alcaraz, M.J. Nrf2 as a therapeutic target for rheumatic diseases. Biochem. Pharmacol. 2018, 152, 338–346.
- Tonelli, C.; Christine Chio, I.I.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97.
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597.
- Kaulmann, A.; Bohn, T. Carotenoids, inflammation, and oxidative stress--implications of cellular signaling pathways and relation to chronic disease prevention. Nutr. Res. 2014, 34, 907–929.
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474.
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-kB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258.
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995.
- Sha, L.K.; Sha, W.; Kuchler, L.; Daiber, A.; Giegerich, A.K.; Weigert, A.; Knape, T.; Snodgrass, R.; Schröder, K.; Brandes, R.P.; et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8(+) T cell function by limiting GSH and Cys availability. Free Radic. Biol. Med. 2015, 83, 77–88.
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598.
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801.
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637.
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione fine-tunes the innate immune response toward antiviral pathways in a macrophage cell line independently of its antioxidant properties. Front. Immunol. 2017, 8, 1239.
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383.
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313.
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554.
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354.
- Ma, L.; Sun, L.; Wang, Y.; Sun, B.; Li, Y.; Jin, Y. Association between HO-1 gene promoter polymorphisms and diseases (Review). Mol. Med. Rep. 2022, 25, 29.
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571.
- Kim, H.P.; Ryter, S.W.; Choi, A.M. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449.
- Zhou, J.; Terluk, M.R.; Basso, L.; Mishra, U.R.; Orchard, P.J.; Cloyd, J.C.; Schröder, H.; Kartha, R.V. N-acetylcysteine provides cytoprotection in murine oligodendrocytes through heme oxygenase-1 activity. Biomedicines 2020, 8, 240.
- Jazwa, A.; Cuadrado, A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets 2010, 11, 1517–1531.
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078.
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the heme oxygenase-1 gene via regulation of Nrf2 and the antioxidant responsive element. Biochem. J. 2003, 371, 887–895.
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029.
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913.
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272.
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259.
- Motterlini, R.; Foresti, R. Heme oxygenase-1 as a target for drug discovery. Antioxid. Redox Signal. 2014, 20, 1810–1826.
- Fagone, P.; Piombino, E.; Mangano, K.; De Pasquale, R.; Nicoletti, F.; Caltabiano, R. Evaluation of the involvement of Heme Oxygenase-1 expression in discoid Lupus Erythematosus lesions. Antioxidants 2023, 12, 1352.
- Li, J.; Stein, T.D.; Johnson, J.A. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genom. 2004, 18, 261–272.
- Bayo Jimenez, M.T.; Frenis, K.; Hahad, O.; Steven, S.; Cohen, G.; Cuadrado, A.; Münzel, T.; Daiber, A. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic. Biol. Med. 2022, 187, 72–91.
- Mangano, K.; Cavalli, E.; Mammana, S.; Basile, M.S.; Caltabiano, R.; Pesce, A.; Puleo, S.; Atanasov, A.G.; Magro, G.; Nicoletti, F.; et al. Involvement of the Nrf2/HO-1/CO axis and therapeutic intervention with the CO-releasing molecule CORM-A1, in a murine model of autoimmune hepatitis. J. Cell. Physiol. 2018, 233, 4156–4165.
- Nikolic, I.; Saksida, T.; Mangano, K.; Vujicic, M.; Stojanovic, I.; Nicoletti, F.; Stosic-Grujicic, S. Pharmacological application of carbon monoxide ameliorates islet-directed autoimmunity in mice via anti-inflammatory and anti-apoptotic effects. Diabetologia 2014, 57, 980–990.
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306.
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381.
- Ahn, K.S.; Aggarwal, B.B. Transcription Factor NF-κB: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218.
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408.
- Ates, I.; Suzen, H.S.; Yucesoy, B.; Ozel Tekin, I.; Karakaya, A. Association of cytokine gene polymorphisms in CWP and its severity in Turkish coal workers. Am. J. Ind. Med. 2008, 51, 741–747.
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117.
- Yan, N.; Xu, Z.; Qu, C.; Zhang, J. Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via NRF2/ARE/NF-κB signal pathway. Int. Immunopharmacol. 2021, 98, 107844.
- Ding, L.; Yuan, X.; Yan, J.; Huang, Y.; Xu, M.; Yang, Z.; Yang, N.; Wang, M.; Zhang, C.; Zhang, L. Nrf2 exerts mixed inflammation and glucose metabolism regulatory effects on murine RAW264.7 macrophages. Int. Immunopharmacol. 2019, 71, 198–204.
- Behl, T.; Kaur, I.; Sehgal, A.; Sharma, E.; Kumar, A.; Grover, M.; Bungau, S. Unfolding Nrf2 in diabetes mellitus. Mol. Biol. Rep. 2021, 48, 927–939.
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotoc ininduced diabetic nephropathy. Diabetes 2011, 60, 3055–3066.
- El-Bab, M.F.; Zaki, N.S.; Mojaddidi, M.A.; Al-Barry, M.; El-Beshbishy, H.A. Diabetic retinopathy is associated with oxidative stress and mitigation of gene expression of antioxidant enzymes. Int. J. Gen. Med. 2013, 6, 799.
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Chorev, M.; Halperin, J.A. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr. Rev. 2015, 36, 272–288.
- Kumagai, Y.; Kanda, H.; Shinkai, Y.; Toyama, T. The role of the Keap1/Nrf2 pathway in the cellular response to methylmercury. Oxidative Med. Cell. Longev. 2013, 2013, 848279.
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47.
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449.
- Cao, Z.; Cooper, M.E. Pathogenesis of diabetic nephropathy. J. Diabetes Investig. 2011, 2, 243–247.
- Seldon, M.P.; Silva, G.; Pejanovic, N.; Larsen, R.; Gregoire, I.P.; Filipe, J.; Anrather, J.; Soares, M.P. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappa B RelA phosphorylation at serine 276. J. Immunol. 2007, 179, 7840–7851.
- Bahadoran, Z.; Tohidi, M.; Nazeri, P.; Mehran, M.; Azizi, F.; Mirmiran, P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: A randomized double-blind clinical trial. Int. J. Food Sci. Nutr. 2012, 63, 767–771.
- Wang, Y.; Zhang, Z.; Sun, W.; Tan, Y.; Liu, Y.; Zheng, Y.; Liu, Q.; Cai, L.; Sun, J. Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxidative Med. Cell. Longev. 2014, 2014, 123963.
- Pereira, A.; Fernandes, R.; Crisostomo, J.; Seiça, R.M.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14357.
- Patel, B.; Mann, G.E.; Chapple, S.J. Concerted redox modulation by sulforaphane alleviates diabetes and cardiometabolic syndrome. Free Radic. Biol. Med. 2018, 122, 150–160.
- Ellison, D.H. Bardoxolone methyl in type 2 diabetes and advanced chronic kidney disease. N. Eng. J. Med. 2014, 370, 1768.
- Tan, S.M.; Sharma, A.; Stefanovic, N.; Yuen, D.Y.C.; Karagiannis, T.C.; Meyer, C.; Ward, K.W.; Cooper, M.E.; de Haan, J.B. Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 2014, 63, 3091–3103.
- Ha, C.M.; Park, S.; Choi, Y.K.; Jeong, J.-Y.; Oh, C.J.; Bae, K.-H.; Lee, S.J.; Kim, J.-H.; Park, K.-G.; Jun, D.Y.; et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vasc. Pharmacol. 2014, 63, 29–36.
- Lazaro, I.; Lopez-Sanz, L.; Bernal, S.; Oguiza, A.; Recio, C.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Madrigal-Matute, J.; Gomez-Guerrero, C. Nrf2 activation provides atheroprotection in diabetic mice through concerted upregulation of antioxidant, anti-inflammatory, and autophagy mechanisms. Front. Pharmacol. 2018, 9, 819.
- Yu, W.; Liu, W.; Xie, D.; Wang, Q.; Xu, C.; Zhao, H.; Lv, J.; He, F.; Chen, B.; Yamamoto, T.; et al. High Level of Uric Acid Promotes Atherosclerosis by Targeting NRF2-Mediated Autophagy Dysfunction and Ferroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 9304383.
- Li, H.; Zhuang, W.; Xiong, T.; Park, W.S.; Zhang, S.; Zha, Y.; Yao, J.; Wang, F.; Yang, Y.; Chen, Y.; et al. Nrf2 deficiency attenuates atherosclerosis by reducing LOX-1-mediated proliferation and migration of vascular smooth muscle cells. Atherosclerosis 2022, 347, 1–16.
- Zhao, Z.; Wang, X.; Zhang, R.; Ma, B.; Niu, S.; Di, X.; Ni, L.; Liu, C. Melatonin attenuates smoking-induced atherosclerosis by activating the Nrf2 pathway via NLRP3 inflammasomes in endothelial cells. Aging 2021, 13, 11363–11380.
- Feng, Z.; Wang, C.; Jin, Y.; Meng, Q.; Wu, J.; Sun, H. Kaempferol-induced GPER upregulation attenuates atherosclerosis via the PI3K/AKT/Nrf2 pathway. Pharm. Biol. 2021, 59, 1106–1116.
This entry is offline, you can click here to edit this entry!