Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Multi-omics is a cutting-edge approach that combines data from different biomolecular levels, such as DNA, RNA, proteins, metabolites, and epigenetic marks, to obtain a holistic view of how living systems work and interact. Multi-omics has been used for various purposes in biomedical research, such as identifying new diseases, discovering new drugs, personalizing treatments, and optimizing therapies.
- OMICs
- data analysis
- cancer
- proteomics
- data integration
1. Introduction
The complexity of biological systems is beyond the scope of single-omics studies, which only focus on one type of biological molecule. To fully understand the molecular mechanisms and interactions that underlie biological functions and diseases, it is necessary to integrate data from multiple omics levels, such as genomics, transcriptomics, proteomics, metabolomics, and epigenomics. This is the essence of multi-omics, an emerging approach that aims to provide a comprehensive and systematic view of biological systems. Multi-omics has been applied to various fields of biomedical research, such as diagnostics, drug discovery, personalized medicine, and synthetic biology. By combining different types of omics data, multi-omics can reveal novel insights into the molecular basis of diseases and drug responses, identify new biomarkers and therapeutic targets, and predict and optimize individualized treatments. Multi-omics has the potential to revolutionize the field of pharmaceutical sciences and enable the development of innovative and effective therapeutics.
However, the multi-omics approach faces many challenges, such as data heterogeneity, integration, analysis, interpretation, and validation. The high dimensionality, diversity, and complexity of multi-omics data pose significant computational and statistical difficulties for data integration and analysis [1]. The biological interpretation and validation of multi-omics results require extensive knowledge of the field of interest and experimental verification.
2. Different Approaches for Multi-Omics Data Integration
There are different approaches and strategies for integrating omics data for drug discovery, depending on the type, quality, and availability of the data, as well as the biological question and hypothesis [2][3] (Figure 1).
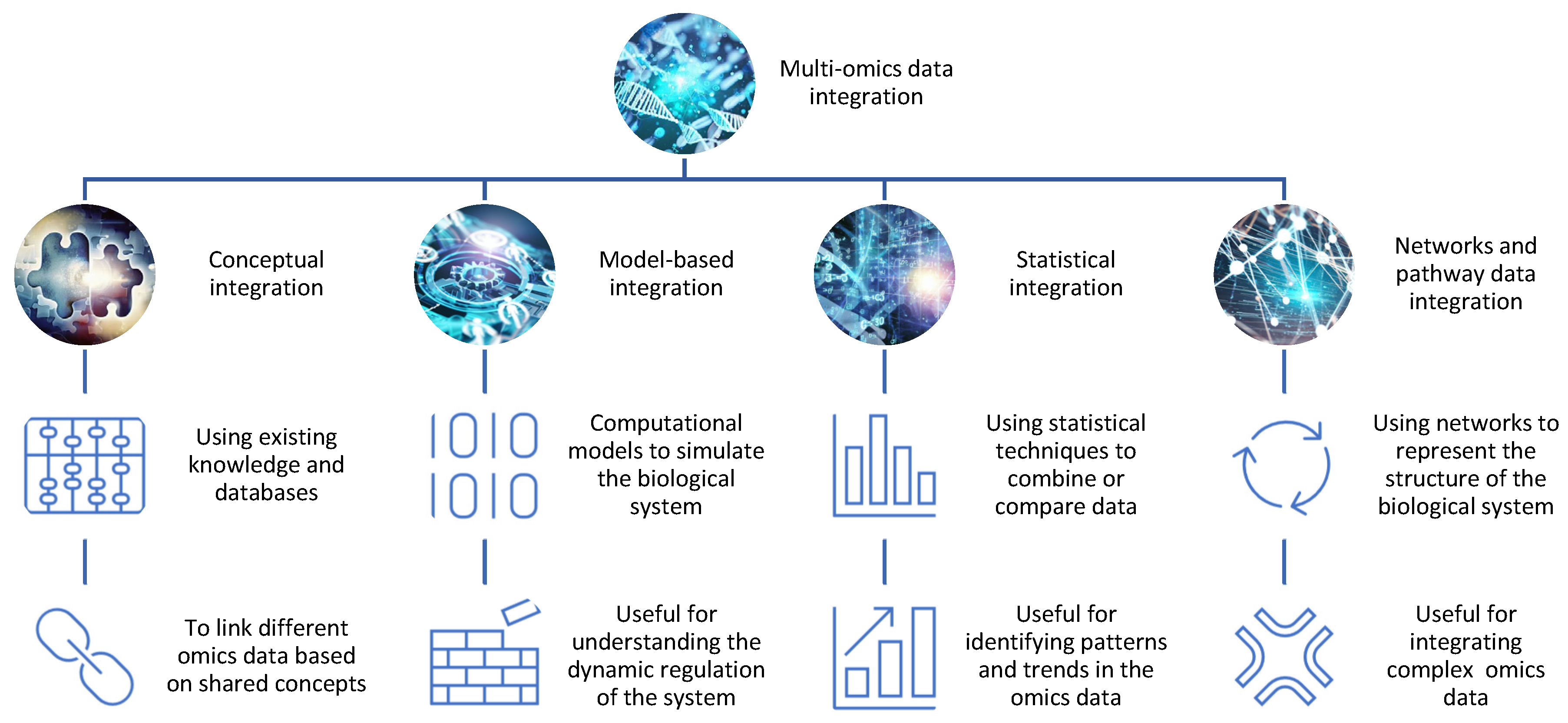
Figure 1. The integration of different omics data for drug discovery. There are different methods and tools for integrating omics data, such as conceptual integration, statistical integration, model-based integration, and network-based integration. Each method has its own advantages and limitations and can reveal distinct aspects of the biological system.
Some of the common methods include:
- −
-
Conceptual integration: This method involves using existing knowledge and databases to link different omics data based on shared concepts or entities, such as genes, proteins, pathways, or diseases. For example, one can use gene ontology (GO) terms or pathway databases to annotate and compare different omics data sets and identify common or specific biological functions or processes [4]. This method is useful for generating hypotheses and exploring associations between different omics data, but it may not capture the complexity and dynamics of the biological system. Open-source pipelines such as STATegra [5] or OmicsON [6] have recently demonstrated an enhanced capacity of the framework to detect specific features overlapping between the compared omics sets;
- −
-
Statistical integration: This method involves using statistical techniques to combine or compare different omics data based on quantitative measures, such as correlation, regression, clustering, or classification [7]. For example, one can use correlation analysis to identify co-expressed genes or proteins across different omics data sets or use regression analysis to model the relationship between gene expression and drug response [8]. This method is useful for identifying patterns and trends in the omics data, but it may not account for the causal or mechanistic relationships between the omics data;
- −
-
Model-based integration: This method involves using mathematical or computational models to simulate or predict the behavior of the biological system based on different omics data [9]. For example, one can use network models to represent the interactions between genes and proteins in different omics datasets or use pharmacokinetic/pharmacodynamic (PK/PD) models to describe the absorption, distribution, metabolism, and excretion (ADME) of drugs in different tissues or organs [10]. This method is useful for understanding the dynamics and regulation of the biological system, but it may require a lot of prior knowledge and assumptions about the system parameters and structure;
- −
-
Networks and pathway data integration: This method involves using networks or pathways to represent the structure and function of the biological system based on different omics data. Networks are graphical representations of the nodes (e.g., genes, proteins) and interactions in the system, while pathways are collections of related biological processes or events that occur in a specific order or context [11]. For example, one can use protein–protein interaction (PPI) networks to visualize the physical interactions between proteins in different omics data sets or use metabolic pathways to illustrate the biochemical reactions involved in drug metabolism [12]. This method is useful for integrating multiple types of omics data at different levels of granularity and complexity, but it may not capture the temporal or spatial aspects of the system.
3. Aims of Multi-Omics Analyses
Data analysis aims to extract useful information or knowledge from omics data that can answer specific research questions or hypotheses (Figure 2). One of the main applications of multi-omics is to identify and validate new drug targets for various diseases. Drug targets are molecules that can be modulated by drugs to alter the disease state or phenotype. Drug targets can be proteins, genes, metabolites, or epigenetic marks that are involved in the pathogenesis or progression of diseases.
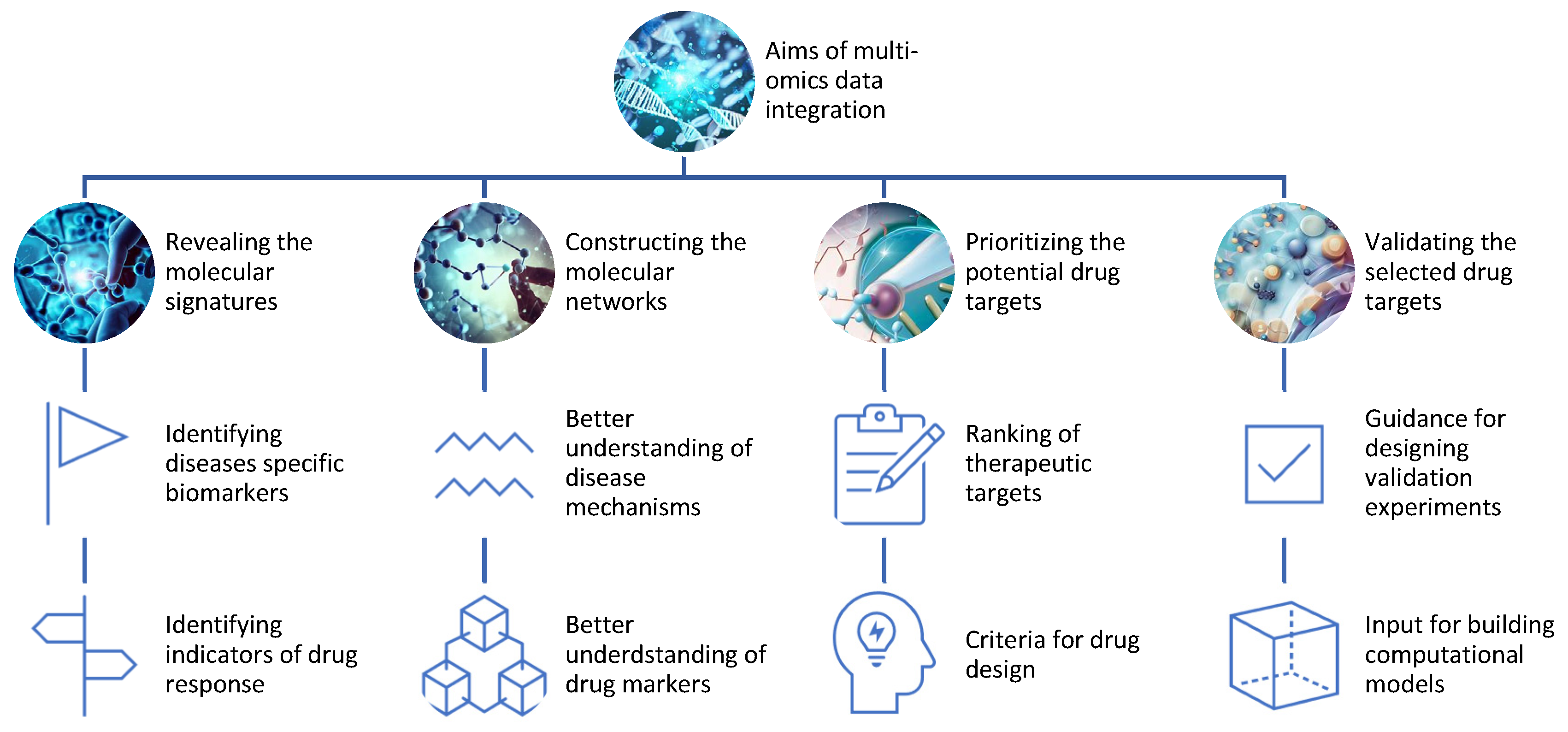
Figure 2. Aims of the integration of different omics data for drug discovery.
Multi-omics can help to discover and validate drug targets by:
- −
-
Revealing the molecular signatures or profiles of diseases and drug responses using omics data from different levels of biological molecules [13]. For example, multi-omics can identify the genes, proteins, metabolites, and epigenetic marks that are differentially expressed or regulated in diseased versus healthy samples or individuals, or in responsive versus non-responsive samples or individuals to a given drug;
- −
-
Constructing the molecular networks or pathways of diseases and drug responses using omics data from different levels of biological molecules [14]. For example, multi-omics can infer the interactions or relationships among genes, proteins, metabolites, and epigenetic marks that are involved in disease mechanisms or drug mechanisms of action;
- −
-
Prioritizing the potential drug targets based on their relevance or importance to diseases and drug responses using omics data from different levels of biological molecules [15]. For example, multi-omics can rank genes, proteins, metabolites, and epigenetic marks based on their differential expression or regulation, network centrality, functional annotation, disease association, drug association, or other criteria;
- −
-
Validating the selected drug targets using experimental methods or computational models that can test the effects of modulating the drug targets on diseases and drug responses. For example, multi-omics can provide guidance for designing experiments such as knockdowns, overexpressions, mutations, inhibitors, activators, or combinations thereof for the drug targets [16]. Alternatively, multi-omics can provide input for building computational models such as PK/PD models, systems pharmacology models, or machine learning models that can simulate the effects of modulating the drug targets [17].
Another main application of multi-omics is to predict and optimize drug responses for various diseases. Drug responses are the outcomes or phenotypes that result from administering drugs to treat diseases. Drug responses can be measured by various indicators such as efficacy, safety, toxicity, adverse effects, resistance, sensitivity, dosage, duration, frequency, or combinations thereof. Multi-omics can help to predict and optimize drug responses by:
- −
-
Characterizing the inter-individual variability of drug responses using omics data from different levels of biological molecules [18]. For example, multi-omics can identify the genetic variants (e.g., single nucleotide polymorphisms (SNPs), copy number variations (CNVs), insertions/deletions (indels)), gene expression levels (e.g., mRNA levels), protein expression levels (e.g., protein levels), metabolite levels, and epigenetic modifications (e.g., DNA methylation levels) that influence how different individuals respond to a given drug;
- −
-
Classifying the subtypes or groups of individuals with similar drug responses using omics data from different levels of biological molecules [19]. For example, multi-omics can cluster individuals based on their molecular signatures or profiles of drug responses into responders versus non-responders, sensitive versus resistant, or toxic versus non-toxic groups;
- −
-
Predicting the optimal drug responses for individual patients using omics data from different levels of biological molecules [20]. For example, multi-omics can use machine learning methods such as SVMs, random forests, or neural networks to build predictive models that can estimate the efficacy, safety, toxicity, adverse effects, resistance, sensitivity, dosage, and duration of drug responses.
4. Different Types of Proteomics Data That Can Be Used for Multi-Omics Analyses
This research, as well as others, highlights the discrepancy between the interactome, proteome, and transcriptome [21]. The discrepancy between interactome/proteome and transcriptome is due to the difference between the levels of transcription of specific genes, translation of mRNA, and protein abundance or interaction in a biological system. This difference can be caused by various factors, such as post-transcriptional regulation, post-translational modification, protein degradation, protein–protein interaction, and environmental stimuli [22]. The discrepancy between interactome/proteome and transcriptome can have significant implications for understanding the molecular mechanisms and functions of biological systems, as well as for identifying potential biomarkers and therapeutic targets for diseases that are linked to protein complexity [23].
Proteome and phosphoproteome are two important concepts in the field of proteomics, which is the study of the entire set of proteins expressed by a cell, tissue, or organism under certain conditions. Proteome refers to the identity, expression levels, and modification of proteins, while phosphoproteome refers to the subset of proteins that are phosphorylated. This is a common post-translational modification that regulates protein function and signaling. Proteomics and phosphoproteomics can provide valuable information for the design of novel therapies [24], especially for diseases such as cancer, where protein expression and phosphorylation are often dysregulated.
Proteomics, ubiquitinome, and phosphoproteomics can also help to characterize molecular mechanisms and target modulators by integrating with other omics data, such as genomics, transcriptomics, and metabolomics. Proteomics can help identify potential biomarkers and protein expression patterns that can be used to assess disease prognosis, tumor classification, and identify potential responders for specific therapies [25]. Phosphoproteomics can help to understand cellular signaling and infer kinase activity, which is a key regulator of many cellular processes and a common target for drug development [26]. In line with these findings, a recent article described an overview of the online data publicly available in the field of cancer research, highlighting the discrepancy between different cancer types and potential multi-omics strategies [27].
The ubiquitinome refers to the set of proteins that are modified by covalently bound ubiquitin molecules, a small protein that regulates protein stability, localization, and function [28]. Ubiquitination is a reversible and dynamic process that can affect various cellular pathways such as the cell cycle, DNA repair, apoptosis, and autophagy. The ubiquitin system is involved in many diseases, such as cardiovascular diseases, cancer, neurodegeneration, inflammation, and infection [29]. Therefore, understanding and manipulating the ubiquitinome, which is the set of all ubiquitinated proteins in a cell or organism, could lead to new therapeutic strategies. One way to use the ubiquitinome for drug discovery is to identify biomarkers or signatures of the ubiquitinome that are associated with certain diseases or conditions. For example, changes in the levels or patterns of ubiquitination of certain proteins can indicate the presence or progression of a disease or the response or resistance to a treatment [30]. By measuring the ubiquitinome using proteomics or other methods, one can diagnose, monitor, or predict the clinical outcomes of patients. Moreover, one can use the information from the ubiquitinome to design novel therapies that target the underlying mechanisms or pathways of the disease, as in the case of Parkinson’s disease (PD) [31][32] or cardiovascular diseases such as Noonan syndrome [33].
Glycoproteome analysis is the study of the structure and function of proteins that are modified by glycans, which are complex carbohydrate chains attached to proteins [34]. Glycoproteins are involved in many biological processes and diseases, such as cell signaling, immune response, cancer, and viral infection. Glycoproteome analysis aims to identify glycoproteins, and their glycosylation sites. Glycosylation can affect the structure, stability, folding, interactions, and functions of proteins and thus regulate many cellular processes and pathways. Moreover, glycosylation can also influence the recognition and response of the immune system to foreign or abnormal cells, such as cancer cells or virus-infected cells. MS-based glycoproteome analysis can be performed using two complementary workflows: glycosylation site mapping and glycopeptide analysis. Glycosylation site mapping identifies the potential glycosylation sites that are occupied by glycans on the protein sequence, while glycopeptide analysis characterizes the specific glycan structures and compositions on each site. Glycoproteome analysis can be used as a powerful tool for disease diagnosis and therapy monitoring because glycosylation can serve as a biomarker that reflects the current status of the patient and the changes in the glycome due to disease progression or treatment [35]. Therefore, analyzing the glycome can reveal the alterations in glycosylation that are associated with different diseases, such as cancer, diabetes, Alzheimer’s disease, and infectious diseases. For example, cancer cells often have abnormal glycosylation patterns that affect their growth, invasion, metastasis, and immune evasion.
Protein acetylation is a type of post-translational modification that involves the addition of an acetyl group to a protein molecule. This modification can affect different amino acid residues of the protein, such as lysine, serine, and threonine. However, the most common and well-studied form of protein acetylation is the acetylation of lysine side chains. Protein acetylation can affect the structure, function, and interactions of proteins and regulate various biological processes such as metabolism and signaling. Protein acetylation can also occur at the N-terminus of the proteins, which is called N-terminal acetylation. N-terminal acetylation is catalyzed by a group of enzymes called N-terminal acetyltransferases (NATs). N-terminal acetylation can affect the protein’s lifetime by influencing its degradation, folding, localization, and interactions with other molecules. For example, N-terminal acetylation can protect proteins from being degraded by proteases that recognize unmodified N-termini, or it can target proteins for degradation by specific ubiquitin ligases. Acetylome analysis is the study of the global patterns and dynamics of protein acetylation using mass spectrometry and bioinformatics tools [36]. Acetylome analysis can reveal critical features of lysine acetylation, such as its abundance, distribution, conservation, and functional roles. Furthermore, it can reveal the changes in protein acetylation patterns and levels that are associated with disease pathogenesis and progression [37]. Acetylome can also be a target for therapy development, such as using drugs that modulate the activity of acetyltransferases or deacetylases, which are enzymes that add or remove acetyl groups from proteins. For example, elamipretide and nicotinamide mononucleotide are mitochondrial-targeted drugs that can restore the abundance and acetylation of proteins that are disrupted by aging in mouse hearts [38].
One way to resolve the discrepancy between the interactome, proteome, and transcriptome is to use multi-omics approaches, which integrate data from different levels of biological molecules. For example, one study used multi-omics data from human brain organoids to identify the posttranscriptional regulation of ribosomal genes by a transcription factor called KLF4 [39]. Another study used multi-omics data from human melanoma samples to identify the molecular mechanisms and therapeutic targets of a novel oncogene called RREB1 [40]. While transcriptomics does not always correlate with protein levels and direct drug responses, the transcriptome can still be important for the design of therapies associated with large chromosomal rearrangements that can, for example, occur in cancer. Several papers have described the integration of RNA sequence, copy number aberration, and methylation to give a better understanding of cellular alterations associated with disease pathogenesis [41]. Such an approach can help identify genes that are affected by multiple types of genomic and epigenomic alterations. A recent article used transcriptome analysis to compare the effects of commonly observed chromosomic deletion on the gene expression in kidney epithelial cells and clear-cell renal cell carcinoma (ccRCC) samples [42].
This entry is adapted from the peer-reviewed paper 10.3390/proteomes11040034
References
- Vahabi, N.; Michailidis, G. Unsupervised Multi-Omics Data Integration Methods: A Comprehensive Review. Front. Genet. 2022, 13, 854752.
- Graw, S.; Chappell, K.; Washam, C.L.; Gies, A.; Bird, J.; Robeson, M.S., 2nd; Byrum, S.D. Multi-omics data integration considerations and study design for biological systems and disease. Mol. Omics 2021, 17, 170–185.
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Brief. Bioinform. 2020, 21, 1937–1953.
- Adossa, N.; Khan, S.; Rytkönen, K.T.; Elo, L.L. Computational strategies for single-cell multi-omics integration. Comput. Struct. Biotechnol. J. 2021, 19, 2588–2596.
- Planell, N.; Lagani, V.; Sebastian-Leon, P.; van der Kloet, F.; Ewing, E.; Karathanasis, N.; Urdangarin, A.; Arozarena, I.; Jagodic, M.; Tsamardinos, I.; et al. STATegra: Multi-Omics Data Integration—A Conceptual Scheme with a Bioinformatics Pipeline. Front. Genet. 2021, 12, 620453.
- Turek, C.; Wróbel, S.; Piwowar, M. OmicsON—Integration of omics data with molecular networks and statistical procedures. PLoS ONE 2020, 15, e0235398.
- Gu, Z.; El Bouhaddani, S.; Pei, J.; Houwing-Duistermaat, J.; Uh, H.W. Statistical integration of two omics datasets using GO2PLS. BMC Bioinform. 2021, 22, 131.
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124.
- ElKarami, B.; Alkhateeb, A.; Qattous, H.; Alshomali, L.; Shahrrava, B. Multi-omics Data Integration Model Based on UMAP Embedding and Convolutional Neural Network. Cancer Inform. 2022, 21, 11769351221124205.
- Li, X.; Ma, J.; Leng, L.; Han, M.; Li, M.; He, F.; Zhu, Y. MoGCN: A Multi-Omics Integration Method Based on Graph Convolutional Network for Cancer Subtype Analysis. Front. Genet. 2022, 13, 806842.
- Liu, X.Y.; Mei, X.Y. Prediction of drug sensitivity based on multi-omics data using deep learning and similarity network fusion approaches. Front. Bioeng. Biotechnol. 2023, 11, 1156372.
- Zhou, G.; Li, S.; Xia, J. Network-Based Approaches for Multi-omics Integration. Methods Mol. Biol. 2020, 2104, 469–487.
- Kreitmaier, P.; Katsoula, G.; Zeggini, E. Insights from multi-omics integration in complex disease primary tissues. Trends Genet. 2023, 39, 46–58.
- Doran, S.; Arif, M.; Lam, S.; Bayraktar, A.; Turkez, H.; Uhlen, M.; Boren, J.; Mardinoglu, A. Multi-omics approaches for revealing the complexity of cardiovascular disease. Brief Bioinform. 2021, 22, bbab061.
- Doherty, L.M.; Mills, C.E.; Boswell, S.A.; Liu, X.; Hoyt, C.T.; Gyori, B.; Buhrlage, S.J.; Sorger, P.K. Integrating multi-omics data reveals function and therapeutic potential of deubiquitinating enzymes. eLife 2022, 11, e72879.
- Li, K.; Du, Y.; Li, L.; Wei, D.Q. Bioinformatics Approaches for Anti-cancer Drug Discovery. Curr. Drug Targets 2020, 21, 3–17.
- Béal, J.; Pantolini, L.; Noël, V.; Barillot, E.; Calzone, L. Personalized logical models to investigate cancer response to BRAF treatments in melanomas and colorectal cancers. PLoS Comput. Biol. 2021, 17, e1007900.
- Allesøe, R.L.; Lundgaard, A.T.; Medina, R.H.; Aguayo-Orozco, A.; Johansen, J.; Nissen, J.N.; Brorsson, C.; Mazzoni, G.; Niu, L.; Biel, J.H.; et al. Discovery of drug-omics associations in type 2 diabetes with generative deep-learning models. Nat. Biotechnol. 2023, 41, 399–408.
- Gallego-Paüls, M.; Hernández-Ferrer, C.; Bustamante, M.; Basagaña, X.; Barrera-Gómez, J.; Lau, C.-H.E.; Siskos, A.P.; Vives-Usano, M.; Ruiz-Arenas, C.; Wright, J.; et al. Variability of multi-omics profiles in a population-based child cohort. BMC Med. 2021, 19, 166.
- Sammut, S.J.; Crispin-Ortuzar, M.; Chin, S.F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629.
- Rao, P.K.; Li, Q. Protein turnover in mycobacterial proteomics. Molecules 2009, 14, 3237–3258.
- Sewduth, R.N.; Baietti, M.F.; Sablina, A.A. Cracking the Monoubiquitin Code of Genetic Diseases. Int. J. Mol. Sci. 2020, 21, 3036.
- Bludau, I.; Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340.
- Gerritsen, J.S.; White, F.M. Phosphoproteomics: A valuable tool for uncovering molecular signaling in cancer cells. Expert Rev. Proteomics 2021, 18, 661–674.
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333.
- Savage, S.R.; Zhang, B. Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin. Proteom. 2020, 17, 27.
- Das, T.; Andrieux, G.; Ahmed, M.; Chakraborty, S. Integration of Online Omics-Data Resources for Cancer Research. Front. Genet. 2020, 11, 578345.
- Swatek, K.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Sewduth, R.N.; Jaspard-Vinassa, B.; Peghaire, C.; Guillabert, A.; Franzl, N.; Larrieu-Lahargue, F.; Moreau, C.; Fruttiger, M.; Dufourcq, P.; Couffinhal, T.; et al. The ubiquitin ligase PDZRN3 is required for vascular morphogenesis through Wnt/planar cell polarity signalling. Nat. Commun. 2014, 5, 4832.
- Du, T.; Song, Y.; Ray, A.; Wan, X.; Yao, Y.; Samur, M.K.; Shen, C.; Penailillo, J.; Sewastianik, T.; Tai, Y.T.; et al. Ubiquitin receptor PSMD4/Rpn10 is a novel therapeutic target in multiple myeloma. Blood 2023, 141, 2599–2614.
- Buneeva, O.; Medvedev, A. Atypical Ubiquitination and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 3705.
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell Proteomics 2013, 12, 3543–3558.
- Sewduth, R.N.; Pandolfi, S.; Steklov, M.; Sheryazdanova, A.; Zhao, P.; Criem, N.; Baietti, M.F.; Lechat, B.; Quarck, R.; Impens, F.; et al. The Noonan Syndrome Gene Lztr1 Controls Cardiovascular Function by Regulating Vesicular Trafficking. Circ. Res. 2020, 126, 1379–1393.
- Shajahan, A.; Heiss, C.; Ishihara, M.; Azadi, P. Glycomic and glycoproteomic analysis of glycoproteins—A tutorial. Anal. Bioanal. Chem. 2017, 409, 4483–4505.
- Van Scherpenzeel, M.; Willems, E.; Lefeber, D.J. Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj. J. 2016, 33, 345–358.
- Guo, J.; Chai, X.; Mei, Y.; Du, J.; Du, H.; Shi, H.; Zhu, J.-K.; Zhang, H. Acetylproteomics analyses reveal critical features of lysine-ε-acetylation in Arabidopsis and a role of 14-3-3 protein acetylation in alkaline response. Stress Biol. 2022, 2, 1.
- Pei, J.; Harakalova, M.; Treibel, T.A.; Lumbers, R.T.; Boukens, B.J.; Efimov, I.R.; van Dinter, J.T.; González, A.; López, B.; El Azzouzi, H.; et al. H3K27ac acetylome signatures reveal the epigenomic reorganization in remodeled non-failing human hearts. Clin. Epigenetics 2020, 12, 106.
- Whitson, J.A.; Johnson, R.; Wang, L.; Bammler, T.K.; Imai, S.-I.; Zhang, H.; Fredrickson, J.; Latorre-Esteves, E.; Bitto, A.; MacCoss, M.J.; et al. Age-related disruption of the proteome and acetylome in mouse hearts is associated with loss of function and attenuated by elamipretide (SS-31) and nicotinamide mononucleotide (NMN) treatment. GeroScience 2022, 44, 1621–1639.
- Sidhaye, J.; Trepte, P.; Sepke, N.; Novatchkova, M.; Schutzbier, M.; Dürnberger, G.; MechtlerJürgen, G.; Knoblich, A. Integrated transcriptome and proteome analysis reveals posttranscriptional regulation of ribosomal genes in human brain organoids. eLife 2023, 12, e85135.
- Schwartz, G.W.; Petrovic, J.; Zhou, Y.; Faryabi, R.B. Differential Integration of Transcriptome and Proteome Identifies Pan-Cancer Prognostic Biomarkers. Front. Genet. 2018, 9, 205.
- Riku, L.; Sampsa, H. CNAmet: An R package for integrating copy number, methylation and expression data. Bioinformatics 2011, 11, 887–888.
- Baietti, M.F.; Zhao, P.; Crowther, J.; Sewduth, R.N.; De Troyer, L.; Debiec-Rychter, M.; Sablina, A.A. Loss of 9p21 Regulatory Hub Promotes Kidney Cancer Progression by Upregulating, H.O.XB13. Mol. Cancer Res. 2021, 19, 979–990.
This entry is offline, you can click here to edit this entry!