However, predicting which patients with SSc-ILD will develop PPF remains a challenge. Various studies have identified several predictors of SSc-ILD progression (i.e., male sex, diffuse cutaneous disease), but translating these observations into clinical practice is difficult, in part because of the different definitions of ILD progression considered in each study (i.e., different thresholds of forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO) decline).
2. Identification of Patients with Progressive Fibrosing SSc-ILD and Diagnosis
A percentage of patients with connective tissue disease (CTD)-ILD develop a PF phenotype, the main features of which are an increase in fibrotic changes in the lung (i.e., traction bronchiectasis and honeycombing) on HRCT, worsening PFT, worsening symptoms, and increased mortality [
1,
4]. Because of the implications for patient counseling and management, it is important to identify the progression of fibrosing ILD as early as possible. In SSc, the variable rates of disease progression and response to treatment emphasize the necessity for close monitoring of all SSc-ILD patients following diagnosis and initiation of treatment [
10,
11,
12].
Many patients with severe SSc-ILD develop this complication within the first 5 years of disease, although some SSc-ILD patients may remain stable for some time and show progression later in the disease course [
13]. In an attempt to identify the different outcomes from different progression patterns and the risk factors associated with the development of PF-ILD, Hoffmann-Vold et al. recently analyzed the presence of progression in 826 patients with SSc-ILD and long-term follow-up, included in the European Scleroderma Trials and Research (EUSTAR) database. Over the course of 12 ± 3 months, 219 (27%) patients exhibited PF ILD with either moderate (FVC decline ranging between 5% and 10%) or significant (FVC decline greater than 10%). In any 12-month time frame, 23% to 27% of patients with SSc-ILD developed PF ILD, although only a minority had a progression in consecutive periods. The most common pattern in patients with progressive ILD (58%) was characterized by a gradual decline in lung function and more frequent periods of stability/improvement compared to decline. On the other hand, about 8% showed the opposite pattern, with a rapid, continuous decline in FVC [
4]. Similarly, recent observational studies have shown that approximately one-third of patients with SSc-ILD experience PPF. In a national Norwegian cohort including 391 patients with SSc-ILD and a mean follow-up period of 6 years, 33% presented with severe progression of ILD, defined as a decline in FVC > 10% of predicted or a decline in FVC between 5 and 10% of predicted, with a decline in DLCO ≥ 15% of predicted [
1].
There are no published clinical practice guidelines for the detection and early diagnosis of ILD-SSc or for monitoring its progression [
14]. The British Society for Rheumatology (BSR)/British Health Professionals in Rheumatology (BHPR) guideline for the management of SSc recommends that all patients with SSc should be assessed for pulmonary fibrosis but does not describe methods of screening or monitoring [
15].
Given the significance of timely recognition and management of ILD, it is crucial to prioritize screening and early diagnosis. It should be noted that SSc-ILD might be present even if there are no respiratory symptoms or a restrictive defect [
1,
16]. It is well known that PFT has very limited sensitivity for the diagnosis of ILD and may be influenced by extra-pulmonary factors that may be present in SSc, such as fatigue, microstomia, severe skin involvement, or myopathy. Nevertheless, PFT remains essential in monitoring the progression of ILD-SSc [
17]. HRCT has been demonstrated to be superior to PFT for the detection of SSc-ILD in the early stages [
16] and is regarded as the gold standard for diagnosing ILD.
Specific laboratory tests may indicate the presence or progression of ILD, including elevated circulating C-reactive protein (CRP) and other acute-phase reactants. Like CRP, elevated serum levels of interleukin (IL)-6 were associated with lung function decline in the first year and with death during the first 30 months of follow-up in an early cohort of SSc-ILD patients [
19]. Although it is possible that IL-6 will play a role in lung fibrosis, both IL-6 and CRP are non-specific for ILD, as their levels can be elevated in almost any inflammatory condition.
Several biomarkers currently under investigation have shown utility in the detection and monitoring of ILD in various diseases. Krebs von den Lungen-6 (KL-6) is a glycoprotein expressed mainly by type II pneumocytes, especially in those cells in the process of proliferation and regeneration. C chemokine ligand 18 (CCL-18), formerly known as lung activation chemokine, is constitutively expressed by lung tissue macrophages and dendritic cells, and it is highly inducible by inflammatory stimulus. It is considered an indicator of lung fibrotic remodeling since it can induce collagen synthesis by lung fibroblasts, thus contributing to lung function deterioration. Serum levels of both proteins have been correlated with the severity of ILD and have been associated with a worse course of this complication in SSc patients [
20]. Similarly, immunosuppressive treatment has been shown to reduce their levels in parallel with the improvement or stabilization of lung function observed in these patients [
21]. Lung epithelial–derived surfactant protein D (SP-D) exerts its function as a component of the innate immune response and plays a role in immune and inflammatory regulation within the lung. In a large European study, including 427 SSc patients, SP-D levels at baseline strongly predicted the presence of ILD but were not associated with the worsening of ILD in longitudinal follow-up [
20]. Despite all these studies, relatively little research has been carried out in this area, and more data in larger longitudinal cohorts is needed to better identify patients at risk for ILD-SSc development and progression.
3. Definition of PPF
Of paramount importance in identifying ILD progression and characterizing clinical phenotypes of PF SSc-ILD is reaching a consensus on how to define progression, given the various definitions that have been proposed and are currently available [
22,
23,
24]. All of these definitions are multidimensional, including symptoms, functional assessments, and imaging to better capture clinically relevant progression.
Cottin et al., 2018 proposed the following criteria for the definition of PF-ILD: a relative decrease in FVC ≥ 10%, a relative decrease in the DLCO ≥ 15%, or a relative decrease in FVC ≥ 5% but <10% combined with worsening of symptoms or radiographic findings in the previous 24 months [
25]. OMERACT (Outcome Measures in Rheumatology) has proposed the definition of “clinically meaningful progression” of CTD-ILD based on the PFT parameters [
26]; this definition can be applied to SSc-ILD, and it is the same as the “ILD progression” proposed by Goh et al. in 2017 [
22]. OMERACT defines progression as a relative decline in FVC ≥ 10%, or a relative decline in FVC ≥ 5% but <10%, associated with a relative decline in the DLCO ≥ 15% over 12 months [
26], which have been identified as surrogate biomarkers for mortality [
4,
13,
23,
24,
25,
27].
In contrast, the eligibility criteria proposed for the INBUILD study (Flaherty et al., 2019), which included ILD of various etiologies other than IPF, all with a progressive pattern, were: a relative decline in FVC ≥ 10%; a relative decline in FVC ≥ 5% but <10% combined with worsening of respiratory symptoms or increased extent of fibrosis on HRCT; or worsening of respiratory symptoms combined with increased extent of fibrosis on HRCT, all within the previous 24 months [
24].
4. Risk Factors for Progressive SSc-ILD
Several studies have attempted to elucidate the most important factors that may predict ILD progression in SSc. Although some factors (i.e., male sex) have been consistently found to predict outcomes in SSc-ILD [
4,
27,
29], other factors have shown inconsistent predictive potential, such as African American race [
30,
31] and plasma CCL18 and CXCL4 levels [
32,
33]. The predictive value of these biomarkers in observational studies and RCTs varies depending on the population studied and on how SSc-ILD progression is defined.
As mentioned above, male sex is independently associated with a higher risk of developing ILD. In a large cohort of 2686 consecutive new SSc patients reported by Peoples et al., males were more likely to have diffuse cutaneous SSc (dcSSc) and ILD, with a significantly reduced survival rate [
27]. In addition, male patients with SSc were significantly more likely to have ever been cigarette smokers and to have environmental exposures. Recently, an analysis of the EUSTAR database was published to determine the impact of gender on outcomes in patients with SSc-ILD. A total of 1136 male and 5253 female patients with SSc-ILD were included. Disease duration and the percentage of predicted FVC in males were associated with greater disease progression. In the survival analysis, male sex was a predictor of mortality [
34].
Patients with an older age at the onset of SSc represent an at-risk subgroup and should be evaluated more frequently for potential organ involvement. In a large German registry of 3281 SSc patients, older dcSSc patients developed lung fibrosis significantly more often (73.5%,
p < 0.001) compared to the younger cohort (55.3%) [
35]. Other demographic factors, such as race, have also been analyzed, and the results showed that ILD is more severe in African Americans compared to European ancestry in SSc patients [
30].
However, the major risk factors for progression of SSc-ILD include diffuse involvement of the skin, the presence of ATA, elevated CRP, worse PFT at baseline, and greater ILD extent on HRCT [
13,
36]. Pulmonary physiology is the best-studied marker of disease progression. Restrictive PFT and impaired gas exchange have consistently been shown to be independent predictors of poor outcomes [
22,
37,
38,
39]. Dynamic monitoring of PFT over time is useful to better predict the course of SSc-ILD. A decrease in FVC > 10%, or of 5–9% with an associated 15% decrease in DLCO, identifies a population at particularly high risk of death [
22]. However, other disease-related factors, such as worsening myopathy, fatigue, or increased skin thickness, should also be taken into account when assessing a decrease in PFT in SSc patients. Cross-sectional imaging has also been the subject of extensive research as a prognostic tool. The initial observation by Goh et al. [
22], that fibrosis involving more than 20% of the lung parenchyma is associated with a significant increase in the odds of mortality, has been confirmed in several independent cohorts [
38,
40]. The presence of a usual interstitial pneumonia (UIP) pattern on imaging or lung pathology is also associated with an aggressive form of ILD [
40].
In addition, other SSc organ manifestations, such as gastroesophageal reflux disease [
41], arthritis, cardiac or renal involvement, digital ulcers, and shorter disease duration, have been associated with the presence of ILD [
36,
38,
39,
42]. In IPF, for example, treatment of gastroesophageal reflux may slow progression [
43]. In SSc, a lack of esophageal contractility is associated with more severe restrictions on PFT [
42]. These findings suggest that the initial assessment of reflux symptoms in SSc-ILD may help to stratify the individual patient risk. Further research is needed to determine how to optimize the treatment of reflux disease in the hope that such treatment may reduce the risk or severity of PPF.
Other biomarkers, such as KL-6, CXCL4, CCL2, CCL18, or surfactant protein-D (SP-D), may predict the progression of SSc-ILD but are not available in clinical practice and are currently used almost exclusively in exploratory clinical research [
13,
32,
44].
Biomolecular profiling via gene expression studies (i.e., exosomes, mitochondrial DNA, microRNA, transcriptomics) may help to identify patients at higher risk of disease progression based on studies developed in cohorts of patients with established SSc. Immune dysregulation, such as upregulated expression of HLA-DRB5, has been reported in patients with ILD-SSc compared with those without ILD [
45]. Telomere shortening also correlates with a poorer outcome in chronic fibrosing ILDs with a PF phenotype, including ILD-SSc [
46]. Furthermore, few studies have investigated epigenetic factors in SSc-ILD patients, including CpG methylation, which is associated with increased DNA methyltransferase expression in fibroblasts [
47].
All of these findings have important clinical implications for the management of SSc-ILD patients, as early treatment is needed for patients at high risk of progression. The approach to identifying ILD-SSc at higher risk of progression represents a paradigm shift in SSc-ILD and highlights the need for reliable and accessible predictive factors for the progression of SSc-ILD, which may help to individualize appropriate and timely follow-up of SSc-ILD patients.
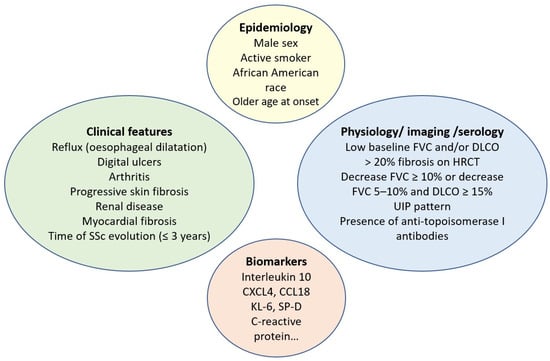
In contrast to ATA, the presence of anti-centromere antibodies (ACA) is associated with a reduced risk of ILD. However, it is important to note that even patients with limited cutaneous SSc and those carrying ACA can develop ILD. It is also known from the serologic profile that anti-RNA polymerase III antibodies are associated with an intermediate risk of ILD [
48]. Risk factors associated with SSc-ILD progression are detailed in
Figure 1.
Figure 1. Risk factors for systemic sclerosis-associated interstitial lung disease progression. SSc: systemic sclerosis; FVC: forced vital capacity; DLCO: diffusing capacity for carbon monoxide; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia; KL-6: Krebs von den Lungen 6; SP-D: surfactant protein-D.