Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Developmental Biology
Congenital heart diseases (CHDs) are structural or functional defects present at birth due to improper heart development. Current therapeutic approaches to treating severe CHDs are primarily palliative surgical interventions during the peri- or prenatal stages, when the heart has fully developed from faulty embryogenesis.
- stem cells
- placenta and heart development
- congenital heart diseases
1. Introduction
The development of the human heart is a complex process that is initiated early during embryogenesis, orchestrated by intricate molecular and cellular events. Congenital heart diseases (CHDs), which result from improper embryonic development, are a group of structural and functional cardiac defects that affect approximately 1% of live births [1]. CHDs may present variable degrees of severity, ranging from minor defects with minimal or no clinical impact to severe malformations requiring immediate medical intervention at birth. Some cardiac conditions may necessitate lifelong treatment and care, frequently resulting in long-term morbidity and increased mortality rates [2]. At present, therapeutic approaches to the treatment of CHDs primarily focus on palliative interventions that improve cardiac structure and function, alleviate symptoms, and reduce the risk of long-term complications. Even surgical interventions, such as cardiac repair or transplantation, which significantly improve survival rates for patients, do not address the underlying genetic abnormalities associated with CHDs. As a result, the risk of CHDs in the children of patients who survived critical CHDs increases to 2–5% [1]. Consequently, there has been a shift from high infant mortality rates to increased survival of adults with complex CHDs, who are at risk for subsequent cardiovascular complications potentially leading to heart failure and the need for lifelong medical care [3]. In the United States, although pediatric admissions for patients with CHDs only account for 3.7% of total admissions, their annual cost is approximately $5.6 billion, which is about 15% of overall costs for pediatric patients [4]. Thus, CHDs are major and increasing health and economic burdens worldwide, and novel strategies to reliably diagnose, refine treatments for, and, if possible, prevent CHDs are urgently needed.
Current prenatal screening programs, genetic counselling, and optimization of maternal health and care, both before and during pregnancy, play a pivotal role in preventing CHDs [5]. The accuracy of prenatal tests to identify cardiac fetal anomalies and the development of innovative medical procedures have opened possibilities for fetal cardiac interventions (FCIs) in utero, at a critical developmental stage when the heart is still growing and developing. FCIs aim to correct fetal cardiac malformations that are either prone to progress with severe complications during mid- or late gestation, or to carry a high risk of fetal demise or life-threatening conditions at birth. FCIs also intend to improve neonatal and prenatal survival of the offspring, and limit lifelong morbidity and mortality [6,7,8,9].
Thus, the limitations of surgical and medicinal treatments of cardiovascular diseases have stimulated the scientific and medical field to search for novel strategies. Consequently, human stem cells have emerged as a promising source of cells for cardiac regeneration in patients to complement current medical and surgical interventions, in addition to serving as cellular models to uncover the underlying mechanisms of CHDs [11,12]. To date, numerous preclinical and clinical studies have been performed to treat CHDs using embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and adult stem cells, such as cardiac progenitor cells and mesenchymal stem cells [13,14,15,16,17]. Although the outcomes of these trials remain largely unsatisfactory, the usage of autologous and allogenic stem cells to complement surgical interventions in infants with CHDs have recently shown positive results, particularly in patients with hypoplastic left heart syndrome (HLHS) [3,13,18,19,20]. To further reduce surgical interventions in patients, the tissue engineering research field is actively developing cell-seeded clinical patches that are able to either grow in synchronization with the cardiovascular structures or to be gradually replaced by the newly formed tissues of treated infants [3]. In addition to their differentiation abilities, stem cells secrete a wide range of bioactive molecules (secretomes) and release extracellular vesicles, including exosomes, which have promising potential in facilitating the repair and regeneration of damaged adult cardiac tissues and improving heart function [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. The secretomes and exosomes released by various cells have significant roles in (cardiac) development and tissue repair, and hold promising potential toward reversing CHDs, or at least mitigating their severity [19,37,38,39,40,41].
The causes of CHDs are often multifactorial and may involve inherited genetic mutations, sporadic developmental errors, and/or environmental factors [42,43,44,45,46]. Mutations or other genomic abnormalities have been shown to occur in genes that play crucial roles in cardiac development, including genes encoding for transcriptional and epigenetic/chromatin remodeling factors (such as NKX2.5, TBX5, GATA4, CITED2, TBX20, p300, and CBP), cell signaling and adhesion proteins (such as ACVR1, NOTCH1, and PDGFRA), and structural sarcomere proteins (such as MYH6, MYH7, and ACTC1) [47,48]. Chromosomal anomalies resulting in syndromic complications, such as Down syndrome (trisomy 21), Turner syndrome (monosomy X), and DiGeorge syndrome (22q11.2 deletion), are often associated with increased risk of CHDs [47,48].
Maternal/embryonic–fetal environmental factors, including maternal diabetes, medications taken during pregnancy, infections, alcohol or drug abuse, exposure to tobacco smoke, chemicals or toxins, may also increase the risk for CHDs [5,48]. Of interest, embryonic heart and placental development are interrelated and concomitant, sharing regulatory molecules and signaling pathways. Placental defects are also more common in pregnancies with CHDs, and the inadequate placental function can contribute to persistent cardiac defects postnatally [50,51,52].
Despite the complexity of embryonic heart formation and the high incidence of CHDs, cardiogenesis is a remarkably robust developmental process. Indeed, dysfunctions of more than one gene and/or environmental insults are often necessary to drastically impair heart development and lead to severe CHDs or death during gestation [53,54,55]. The resilience of this process certainly relies on the complex and evolutionary conserved gene regulatory network, which is orchestrated by key signaling pathways and transcription factors, that belong to families of proteins sometimes displaying overlapping or redundant functions [56]. Their robustness also depends on the existence of multiple sources of cardiac progenitors with functional redundancy, ensuring the production of heart cells even in the event of loss or defects in formation, expansion, and differentiation of certain progenitors [57,58,59]. For instance, despite ablation of over 50% of emerging First Heart Field (FHF) and Secondary Heart Field (SHF) cardiac progenitors at early stages of cardiogenesis, mouse embryos survived and developed into adult animals without any apparent cardiac abnormalities [58,60]. Therefore, the dysfunction or loss of cardiac progenitors and cardiomyocytes in mouse embryonic hearts can truly be compensated by alternative embryonic mechanisms, which include the expansion and migration of other unaffected cardiac cells [58,60,61,62,63].
Recent studies, using animal models and in vitro differentiation of human pluripotent stem cells, have also suggested that cardiac cell lineages are pre-determined during the early stages of development. Both positioning and functions of cardiac cells in the heart may be defined even before gastrulation, and orchestrated by multiple signaling pathways and environmental cues [57,64,65,66,67,68,69]. This idea is also supported by the ability of pluripotent stem cells to generate self-organizing cardiac organoids when provided with external cues from the extracellular matrix (ECM) and signaling molecules [70]. Moreover, in the absence of maternal tissues in vitro, human blastocysts attached to instructive supports and/or, stimulated by signaling molecules (such as WNT and NODAL/ACTIVIN), autonomously self-organize and originate structures resembling the proper embryo, with key landmarks of normal development, including bilaminar disc formation, primitive streak formation, lineage commitment, and extraembryonic annexes [71,72,73,74]. Therefore, future clinical strategies that would consistently provide correct developmental signals at early embryonic stages have the potential to compensate for defective mechanisms, thereby significantly reducing the incidence of CHDs.
2. Navigating Early Mammalian Embryonic Heart and Placental Development
2.1. Placental Development
Placental and embryonic heart developments occur simultaneously, forming a placenta–heart axis, and sharing developmental pathways and common susceptibility to genetic defects [51]. Thus, the developing heart is highly vulnerable to early placental insufficiency. During early human embryogenesis, at embryonic days 4 to 5 (E4-5), the blastocyst attaches to the uterine wall. Trophoblast cells, encasing the inner cell mass that will originate the embryo proper, differentiate into inner cytotrophoblast cells, which are key for blastocyst implantation in the uterus, and stem cells that will also originate the outer syncytiotrophoblast [75,76]. The syncytiotrophoblast is responsible for the exchange of nutrients, gases, and waste products between the maternal and embryonic circulations, and also acts as a barrier against pathogens and harmful substances [77]. Interactions between the cytotrophoblast and syncytiotrophoblast are essential for proper placental development and function. Aberrant vascular placental development can lead to placental insufficiency and compromise fetal growth due to inefficient blood supply, and may result in preeclampsia, gestational diabetes, intrauterine growth restriction (IUGR), and placental abruption [78,79,80,81,82]. These complications are associated with an increased risk of adverse fetal outcomes, including CHDs [83,84,85,86]. Vascular endothelial growth factor (VEGF) and its receptors, hypoxia-inducible factors (HIFs), which regulate the cellular response to hypoxia, play a crucial role in placental and heart development, as well as in vascular remodeling, and their dysfunction increases the risk for CHDs [75,87,88,89]. Immune modulation is also essential for placental development and for fetal tolerance by the maternal immune system [90,91].
2.2. Heart Development
During embryogenesis, the heart is progressively built with multiple mature and functional cells, originating from many molecularly distinct progenitor cells that arise at different times and structures from the developing embryo [57,65,66,67,92,93]. The heart primarily develops from three main pools of embryonic progenitors, which are the FHF, SHF, and the proepicardium cells (Figure 1). The FHF and SHF derive from the earliest MESP1-marked cardiac mesodermal progenitors, which emerge shortly after gastrulation [57,64,65,66,67]. However, precardiac progenitor cells in the nascent mesoderm are, in fact, a heterogeneous population, composed of molecularly distinct progenitors that leave the primitive streak in a sequential manner, with contributions to specific and overlapping parts of the heart [93,94,95].
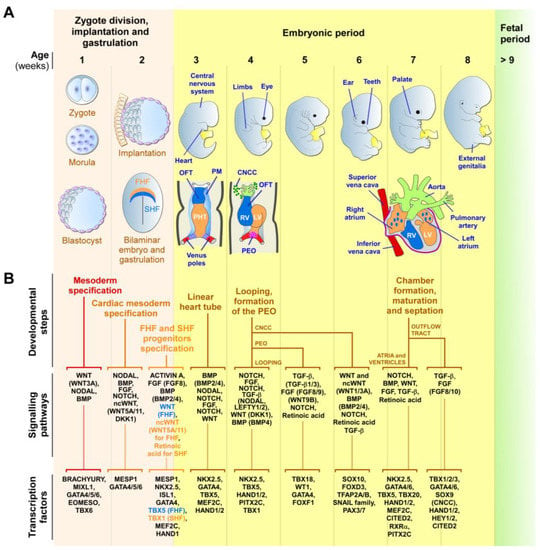
Figure 1. Early human cardiac development. (A) Schematic representation of the zygotic and embryonic stages of human development, highlighting key developmental programs and milestones. Gestational times are indicated in weeks. The initial steps of heart, central nervous system, limb, eye, ear, tooth, and genital development are shown. The primary (FHF—blue) and secondary (SHF—orange) heart field cells, the cardiac neural crest cells (CNCCs—green), and the proepicardial organ (PEO—purple), and their respective derivatives, are illustrated. During week 2, the cardiac mesoderm gives rise to progenitors of the FHF and SHF, which form the cardiac crescent. The FHF originates the primary heart tube (PHT), which subsequently contributes to the left ventricle (LV) and parts of the right and left atria. SHF cells migrate through the pharyngeal mesoderm (PM) and contribute to the elongation of the PHT by ingression at both atrial and venous poles. SHF cells contribute to the development of the right ventricle (RV), outflow tract (OFT), atria, and inflow myocardium. Cells originating from the venous poles (red) give rise to the superior and inferior vena cava. The PEO cells contribute to the epicardium and coronary vessels. CNCCs migrate from the dorsal neural tube into the cardiac OFT, where they contribute to the formation of the septum, separating the truncus arteriosus into the aorta and pulmonary artery, as well as contributing to heart valve formation and parasympathetic innervation. (B) Overview of the secretomes, main signaling pathways, and transcription factors that regulate each listed stage of heart development. The noncanonical WNT pathway is indicated as ncWNT.
The first cells to leave the primitive streak, emerging at approximately E15-16 in humans, are FHF cells that give rise to the cardiac crescent structure, which migrates and fuses at the midline to form the primitive linear heart tube (at around E19 in humans) [42,57,64,65,66,93,96,97]. The linear heart tube, destined to generate the bulk of the atrial chambers and left ventricle, contains an outer myocardium and an inner endocardium, separated by a specific ECM known as the cardiac jelly. Rhythmic contractions are initiated by cardiomyocytes of the heart tube at around E21 in humans. At approximately E19-20, human SHF cells migrate into the heart tube and differentiate into cardiomyocytes, smooth muscle, and endothelial cells. The SHF contributes to the outflow tract (OFT), the right ventricular region, the atrioventricular canal structures, and the atria [42,57,64,65,66,93,96,97].
Around E28, the human heart tube undergoes a rightward looping to initiate the formation of the four-chambered heart. At this stage, the conductive system also begins to develop from cardiac progenitors, which differentiate into specialized myocytes with high conductance to form the sinoatrial node, atrioventricular node, bundle of His, and Purkinje fibers, rather than working cardiomyocytes [98,99]. As the heart develops, conduction cells establish connections to form a functional electrical system that coordinates the contraction and relaxation of the cardiac muscle. Further maturation and refinements of the conductive system occur throughout fetal development, with further structural and functional changes happening after birth and during postnatal growth [98,99]. In humans, ventricular septation starts around E50 and originates from the myocardium, which generates the ventricular septum and establishes the right and left atrio-ventricular canals. Atrial septation starts at E60 and derives from the septum primum and septum secundum, concomitantly with OFT septation [42,67,100,101].
OFT septation, which results in the formation of the aorta and the pulmonary artery, and their respective connections to the left and right ventricles, is accomplished by SHF cells and cardiac neural crest cells (CNCCs), a subpopulation of neural crest cells that delaminate from the neural tube [101,102]. CNCCs also give rise to the tip of the interventricular septum and OFT cushions, which will differentiate into the aortic and pulmonary valves and the parasympathetic coronary innervation [102]. The proepicardial organ is an extracardiac structure, which develops distinctly from the heart tube (at E9.5 in mouse) and contributes to the epicardial cells located around all heart chambers [95,103,104]. Other early multipotent progenitor cells, marked by KDR/FLK1/VEGFR2 expression, which are a source for endocardium, myocardium, and hematopoietic progenitors, were also detected in the primitive streak [57,105]. The major functional structures of the human heart are completed by E60. After this stage, the heart undergoes progressive growth, as well as structural, metabolic, and functional maturation processes, which are vital for its function during a lifetime [70].
3. Unveiling the Potential of Secretomes and Exosomes for CHD Prevention
3.1. Exogenous Instructive Molecules to Mitigate CHDs
Multiple signaling pathways, which may display overlapping or redundant functions, are involved in generating cardiac progenitors and other cell lineages [57,64,65,66,67,68,69]. All issues considered, it is tempting to propose that the supplementation of exogenous cardiogenic instructive molecules at key moments during early embryonic stages may compensate for defective pathways and/or lineage decisions of cardiovascular progenitors to mitigate CHDs. To date, only a few experimental reports on animal models and stem cells support this idea. For instance, prolonged hypoxia causes DNA damage, premature senescence, impaired angiogenesis, and fibrosis associated with the upregulation of TGF-β1 expression in human fetuses with HLHS [201]. In vitro, hypoxia exposure of a human HLHS-derived iPSC disease cell model promotes the differentiation into cardiac fibroblasts instead of cardiac progenitors, cardiomyocytes, and endothelial cells [201]. Inhibition of TGF-β1 activity by its antagonist compound SB431542, in HLHS-derived iPSC exposed to hypoxia, prevented senescence and promoted genomic stability [201], suggesting that early interventions to inhibit TGF-β1 could improve ventricular growth and overcome pathways dysregulated in HLHS. IUGR is another gestational defect that hinders fetal growth in the uterus. IUGR is associated with increased placental vascular resistance, which forces the workload onto the fetal heart and increases the risk of cardiovascular disease [202]. Maternal administration of IGF-1 and IGF-2, which are known to stimulate placental and fetal growth in animal models, showed promising potential to treat IUGR in cases where downregulation of IGF-1 receptors in the placenta is not observed [202,203].
3.2. Exosomes, a Non-Cellular Approach for Correcting Embryonic Cardiac Defects
Emerging evidence suggests that the beneficial effects of stem cells in medical applications may primarily result from paracrine signaling molecules and the release of extracellular vesicles, rather than from the direct integration of stem cells or their derived cells into the target tissue [204]. Exosomes, which can be isolated and characterized by well-established approaches [205,206], present advantages such as scalable production, easy storage, consistent morphology and function, compliance with regulatory standards, and possible reduction in the variability of outcomes associated with cell therapies [207]. The therapeutic potential of exosomes and secretomes derived from hormonally primed human endometrial epithelial cells was tested in mouse models, and proven to enhance embryonic growth, development, and implantation [181]. This study also suggested that dysfunctions of the endometrial secretomes, which hinder implantation in cases of infertility, can be amended through the supply of exogenous exosomes. Also, maternal diabetic pregnancies in mouse models showed a higher occurrence of neural tube defects associated with exosomes produced from vascular progenitor cells expressing FLK1 derived from mesoderm cells lacking Survivin [208]. Interestingly, delivery into the amniotic cavity of Survivin-enriched vascular progenitor exosomes prevented neural tube defects in diabetic pregnancies [208], demonstrating the capacity of modified exosomes delivered in utero to limit neuronal pathology. Thus, exosomes are a promising non-cellular alternative to cells for clinical applications to correct embryonic defects. Exosomes can also be engineered to carry specific cargo, such as miRNAs or even gene-editing tools more suitable for the correction of genetic abnormalities associated with CHDs [176,209]. Notably, exosomes derived from the cardiac progenitors of children undergoing reconstructive heart surgeries showed promising effects in promoting angiogenesis, reducing fibrosis, resolving hypertrophy, and improving cardiac function in a rat model of heart arrhythmias caused by ischemia-reperfusion injury [39], showing the cardioprotective effect of exosomes from infant cardiac cells. Interestingly, exosomes from neonatal progenitors improved cardiac function regardless of oxygen levels, while exosomes from older children were only reparative in hypoxic conditions. Therefore, further research is needed to determine the most suitable cell sources and culture conditions for the production and modification of exosomes that would yield the best personalized clinical outcomes in the treatment of CHDs [40,208].
4. Harnessing Secreted Factors in Embryogenesis for Protection against CHDs
Like in many diseases, the early correction of a CHD is likely to result in a better clinical outcome, through early normalization of cardiogenesis. Reports on stem cell differentiation and heart development have suggested that cell positioning and functions within the heart may be determined prior to gastrulation, through precise signaling pathways and environmental cues [57,64,65,66,67,68,69]. Therefore, early interventions aiming to target the blastocyst at the pre-gastrulation/pre-implantation stage, very early embryonic states, may reduce the occurrence and severity of CHDs. Interestingly, pioneering studies on Inhibitors of Differentiation (ID) genes have revealed that double knockout of any pair of these genes in mouse models results in prominent cardiac defects and midgestational lethality of the embryos [210,211]. However, the injection of wild-type ESCs, either in ID-null blastocysts to form a chimera, or intraperitoneally in females prior to conception, rescued cardiac malformations and viability of ID-null embryos through upregulation of IGF-1 and WNT5A, without incorporation of wild-type ESCs into ID-null embryos [210,211]. Similarly, a chimeric embryo formation, using wild-type ESCs, improved the morphogenetic defects of RA signaling-deficient embryos by increasing RA expression [196]. Interestingly, transient exogenous WNT5A and WNT11, supplied at the 1-cell stage, efficiently rescued the viability and cardiac defects of CITED2-depleted zebrafish embryos and cardiac differentiation of mouse ESCs [212]. Dysfunctions of CITED2, a transcriptional modulator, have been widely associated with zebrafish, mouse, and human CHDs, as well as with embryonic lethality [55,212,213,214,215,216,217,218,219,220]. At the cellular level, CITED2 regulates the expressions of numerous genes involved in early cardiogenesis, such as BRACHYURY, MESP1, ISL1, GATA4, TBX5, MEF2C, NODAL, LEFTY1/2, PITX2C, VEGFA, WNT5A, and WNT11, among others [212,213,214]. Thus, the supplementation of WNT5A and WNT11 at the blastocyst stage in utero holds potential for rescuing CHDs associated with CITED2 dysfunction in mammals, since the function of CITED2 is conserved across vertebrates [212,214,216,221]. This strategy may also prevent CHDs triggered by the dysfunction of many genes other than CITED2, such as CITED2-target cardiogenic genes, including WNT5A and WNT11.
Moreover, WNT5A and WNT11 trigger many congenital cardiac anomalies, and contribute to DiGeorge syndrome, when defective [222,223,224,225,226,227,228]. Indeed, the WNT5A and WNT11 proteins are central for proper gestation and successful pregnancy, and their expressions are naturally highly increased and localized in the uterine luminal epithelium prior to and during blastocyst attachment to the uterus [229,230]. These proteins also promote embryonic uterine implantation and survival [229], as well as placental growth [230]. WNT5A and WNT11 are also important in late gastrulation, for the regulation of the anterior–posterior axis elongation, notochord extension, and proper patterning of the neural tube and somites [231]. In early mouse gastrulation, WNT11 is initially expressed in endoderm progenitors, and later, during mid-gastrulation, it plays a role in the formation of the embryonic and extraembryonic endothelia, as well as the formation of the endocardium in all chambers of the developing heart [232]. During late gastrulation, WNT11 showed successive waves of expression in different regions of the myocardium, important to originate left ventricle precursors (FHF progenitors) from E7.0–8.0, right ventricle progenitors (SHF progenitors) from E8.0–9.0, and the superior wall of the OFT from E8.5–10.5 (also SHF progenitors) [232]. Other studies have implicated WNT5A and WNT11 in the development of cardiac progenitors in vitro and in vivo [233,234,235,236], proper fetal hematopoiesis [237,238], kidney development [239], and guidance of sympathetic neurons to their innervation targets in vivo [240], among other processes. Altogether, the broad range of effects exhibited by WNT5A and WNT11 during early development emphasizes their high potential in limiting CHDs and other birth abnormalities when supplied exogenously at early embryonic stages. Another recent study highlighted the role of a subset of human amniotic epithelial cells (AECs) in mesoderm formation at E8.0–9.0 during early post-implantation stages [241]. Interestingly, impaired mesoderm formation and lethality due to loss of ISL1 expression in non-human primate embryos, or in human AEC differentiation in vitro due to the decrease in BMP4 expression, was partially restored through supplementation of BMP4 [241]. Overall, these findings demonstrate the potential for biomolecules, such as WNT5A/WNT11, BMP4, and inhibitors of the TGF-β pathway (SB431542, for instance), to reduce CHDs when administered at very early stages of development.
This entry is adapted from the peer-reviewed paper 10.3390/jpm13081263
This entry is offline, you can click here to edit this entry!